Two new preprints dealing with the analysis of excited-state electronic wavefunctions are available from ChemRxiv.
First, a quick summary of the TheoDORE package: TheoDORE: a Toolbox for a Detailed and Automated Analysis of Electronic Excited State Computations.
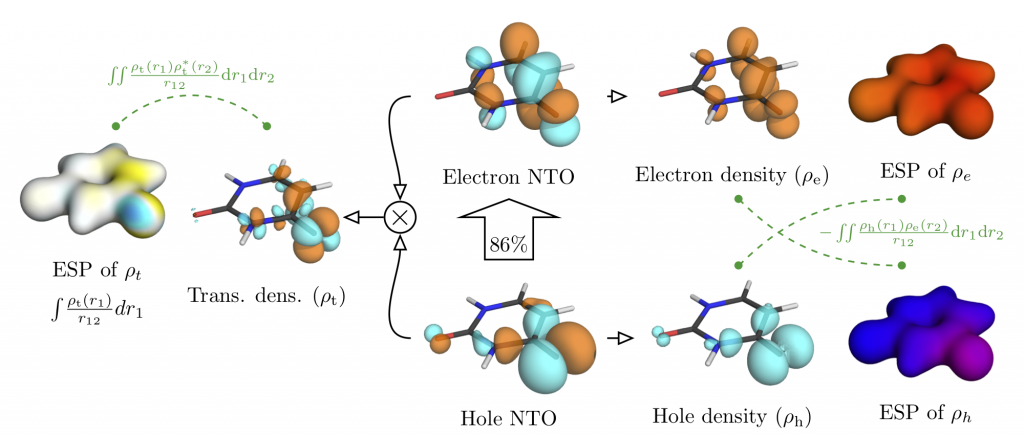
Second, a more extensive paper exploring how far we can use information from excited-state wavefunction analysis tools to understand excitation energies beyond the molecular orbital picture. The energy of a correlated electron-hole pair is derived using diagrammatic techniques and this information is further used for a graphical depiction in terms of different charge distributions and their electrostatic potentials. Doing so turned out not as easy as hoped for but was very exciting. Find more here: Toward an Understanding of Electronic Excitation Energies Beyond the Molecular Orbital Picture by P. Kimber and F. Plasser.