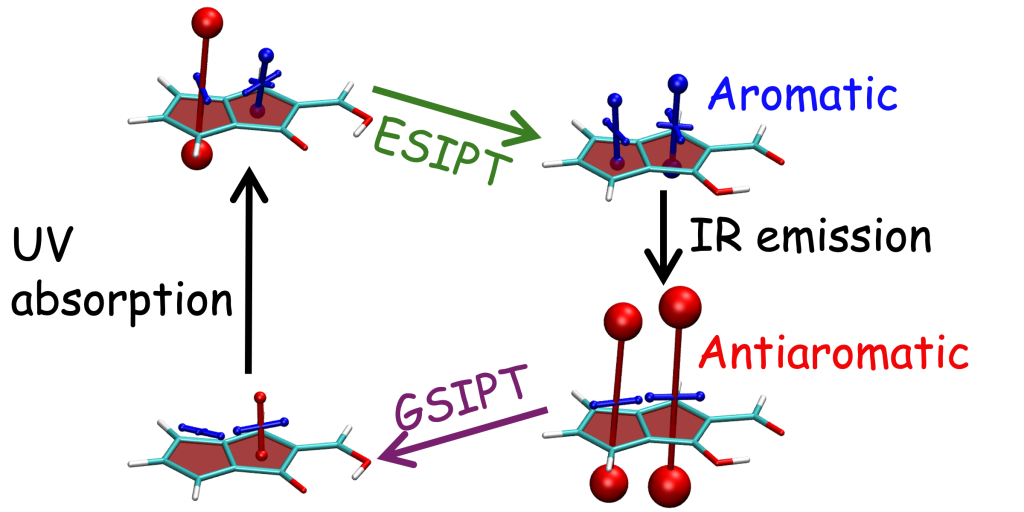
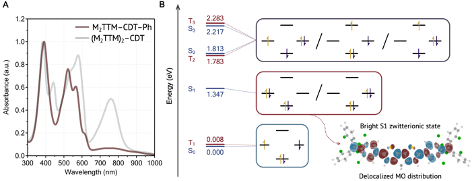
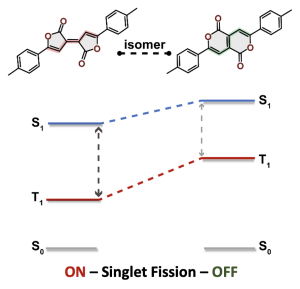
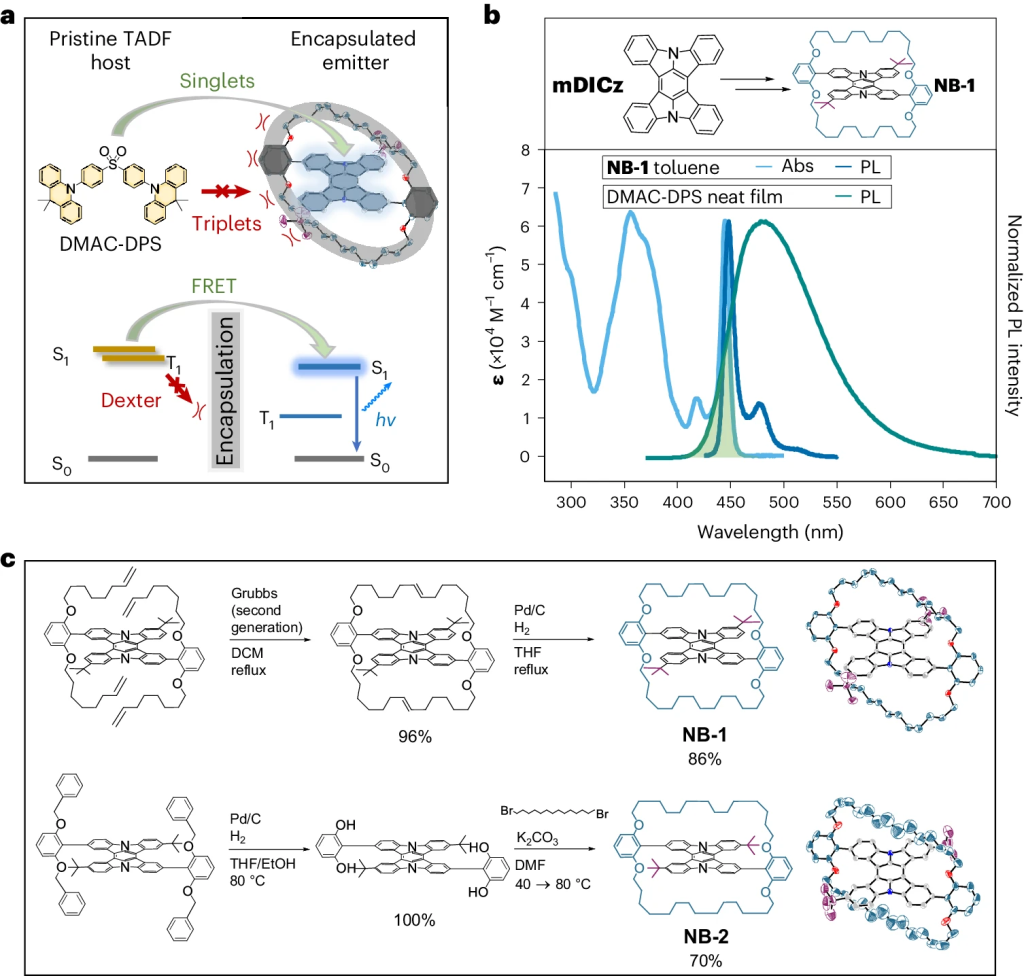
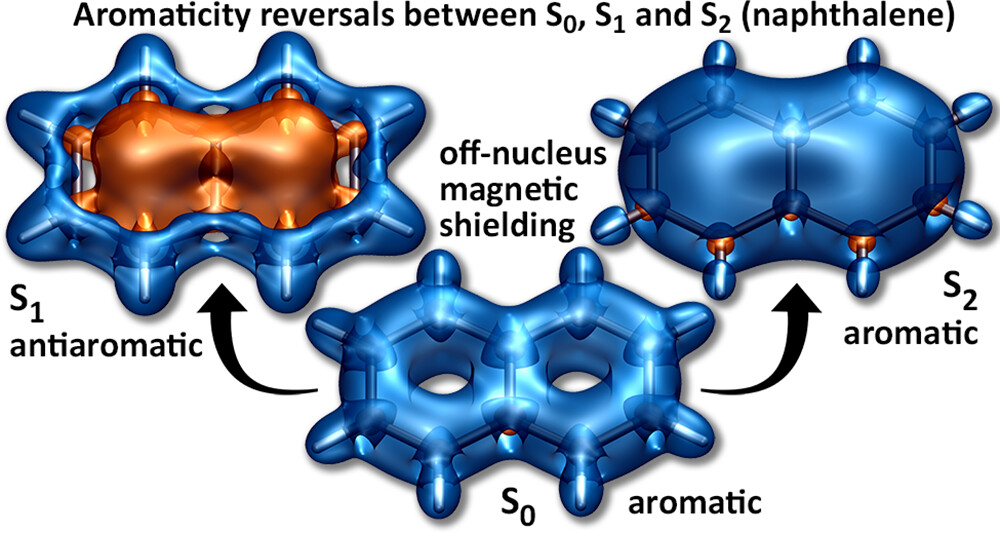
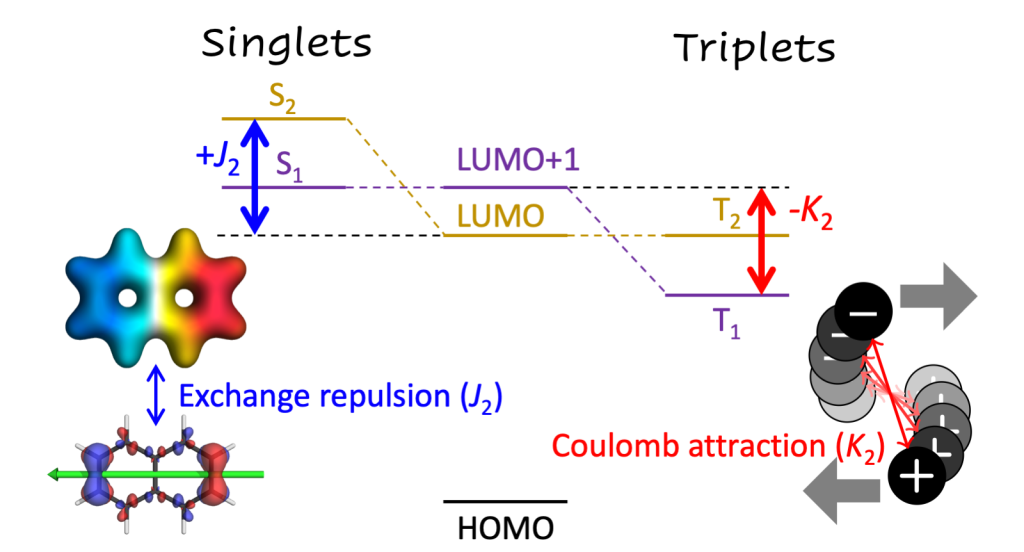
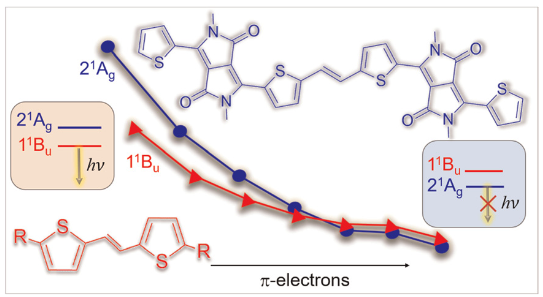
Understanding exciton interactions via a dipole model is often not very intuitive and, more importantly, a dipole model cannot explain the short-range interactions that are often crucial in determining interchromophore interactions.
In our new article [1], exciton coupling in organic chromophores is revisited through the lens of the transition density. The presented formalism gives insight into the strength and sign of the coupling based on the relative arrangement of the lobes of the transition density explaining oscillations between H- and J-aggregate behavior observed when two molecules are displaced relative to each other.

[1] J. Krieger, F. Plasser: Rationalising Exciton Interactions in Aggregates Based on the Transition Density, Chem. Eur. J. 2025, DOI: 10.1002/chem.202501570.