Patrick’s first paper as first author just appeared in PCCP: The role of excited-state character, structural relaxation, and symmetry breaking in enabling delayed fluorescence activity in push-pull chromophores. Well done Patrick!
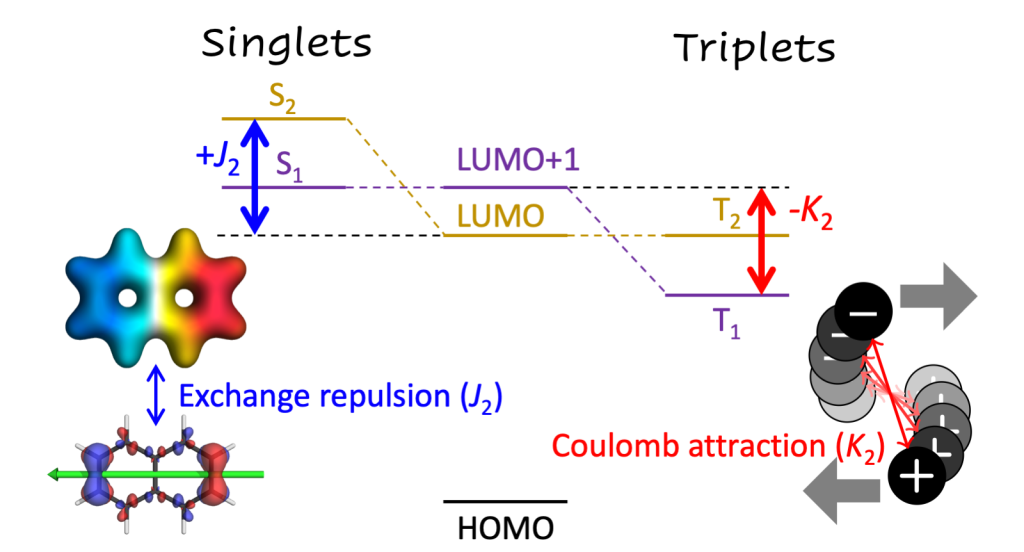
We were interested in understanding the difference in thermally activated delayed fluorescence (TADF) between two closely related donor-acceptor-donor systems using either an anthraquinone and benzodithiophenedione acceptor units, respectively. The first one was known to be an effective TADF emitter [JACS 2014, 136, 18070] whereas the second one had significantly lower quantum yield for TADF [PCCP 2019, 21, 10580].
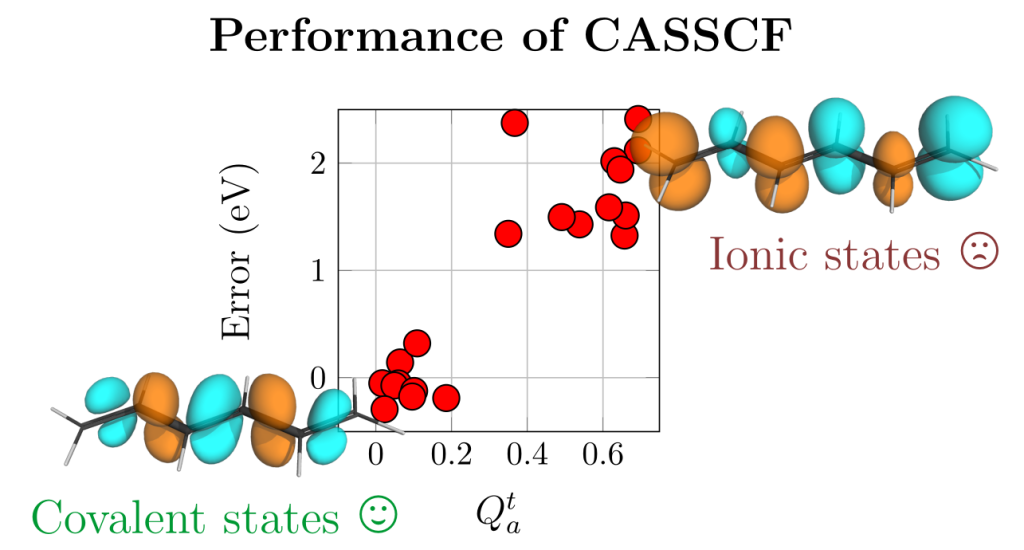
Rather than just presenting energies, it was the purpose of this paper to shed detailed insight into the wavefunctions involved. Notable differences in the wavefunctions and charge-transfer character were found between the two molecules. Even more striking differences existed between different computational methods.
After evaluating electronic structure methods, we presented geometry optimisations in solution, highlighting the importance of symmetry breaking for producing an emissive lowest singlet state. The role of different solvation models was discussed as well.