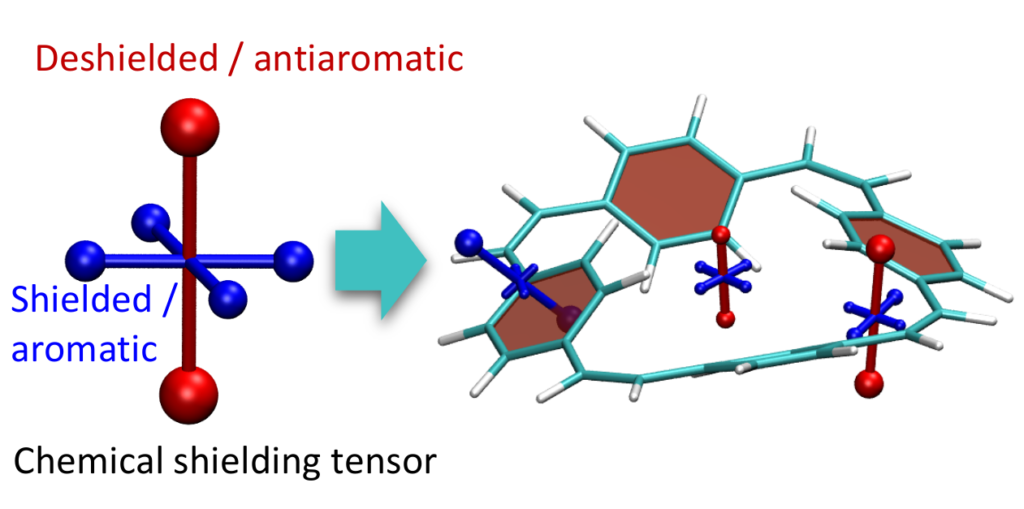
Aromaticity is a ubiquitous yet elusive concept in chemistry and chemists have spent a great deal of effort on developing methods to quantify and visualise aromaticity. One particularly popular method is the nucleus independent shift (NICS), which can be seen as a virtual NMR experiment carried out within a conjugated ring to evaluate the enhanced chemical shielding induced by aromatic ring-currents. Strikingly NICS also allows to quantify antiaromaticity, as this induces a net deshielding effect within the ring. NICS provides a powerful quantitative aromaticity criterion but the main challenge for its graphical representation is that the chemical shielding is a 3×3 tensor, which is difficult to visualise with the existing methods.
Therefore, we have developed a new method for the visualisation of chemical shielding tensors (VIST), which provides a local representation of the shielding tensor along with the molecular structure. The method, thus, allows to probe local aromaticity along with the underlying anisotropy of the shielding. The method is described in the preprint “3D Visualisation of chemical shielding tensors to elucidate aromaticity and antiaromaticity” available on ChemRxiv.
Within the preprent we exemplify the main concepts in the benzene and phenanthrene molecules and continue by studying
the interplay of ground state antiaromaticity and Baird triplet state aromaticity in the potential singlet fission chromophore cyclobuta[l]phenanthrene,
local aromaticity in the neutral formally antiaromatic ground state along with global aromaticity in the doubly reduced state of paaracyclophanetetraene,
A new release of Columbus (version 7.0.1) is available. The main improvement compared to previous releases is that the binary distribution now contains a pre-compiled parallel CI executable as well as an interface to Molcas.
The parallel CI executable is linked against the Intel MPI libraries, which should be available on most computing centres and can otherwise be downloaded freely from Intel.
The Molcas interface proceeds via the free Molcas@UU distribution. Note: At the moment, there is no interface to OpenMolcas available in the distribution.
Source and binaries as well as more detailed instructions are available via the usual download page (register here). Let me know about any successes or problems regarding these features.
Today at 6pm, Felix will give a talk as part of the Q-Chem webinar series. The talk will discuss Q-Chem’s features for extended excited-state wavefunction analysis implemented via the libwfa library.
New Analysis Tools for Excited-State Quantum Chemistry: The libwfa Library in Q-Chem
Tomorrow, Felix will give a seminar talk at the online OpenMolcas meeting. Title: Wavefunction analysis in OpenMolcas: Fragment-based analysis and de-excitations
Columbus is a collection of programs for high-level ab initio electronic structure computations. Through the use of multireference methods even highly challenging systems such as excited states and open-shell molecules are accessible. The availability of gradients and nonadiabatic coupling vectors allows for photodynamics simulations describing ultrafast internal conversion processes. The capabilities of Columbus have been showcased in a recent paper: The generality of the GUGA MRCI approach in COLUMBUS for treating complex quantum chemistry that just appeared in J. Chem. Phys. as part of a themed collection Electronic Structure Software.
A new release of the programm package, available to registered users, has been made available on the distribution page.
In the tutorial, we explain the process of creating conditional electron densities for visualising electron correlation (ChemPhotoChem2019, 3, 702). The figure below shows a comparison between the ionic and covalent singlet and triplet B3u states of naphthalene.
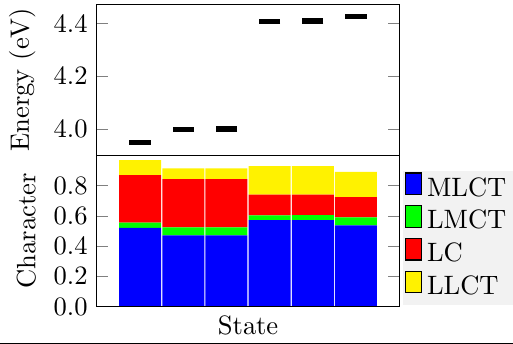
The tutorial also explains the creation of bar graphs for a compact representation of excited-state character (see Coord. Chem. Rev., 2018, 361, 74 and ChemRxiv.11395314). In the picture below, the excited states of an iridium complex are decomposed into metal-to-ligand charge transfer (MLCT), ligand-to-ligand charge transfer (LLCT), and ligand centred (LC) contributions. The lowest six states are all dominated by MLCT character but the presented analysis clearly shows that the first three have enhanced LC character compared to the latter three.
Privacy & Cookies: This site uses cookies. By continuing to use this website, you agree to their use.
To find out more, including how to control cookies, see here:
Cookie Policy