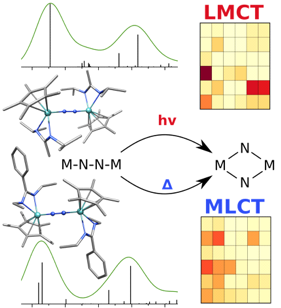
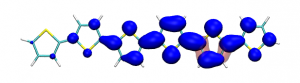
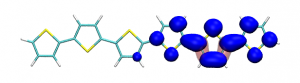
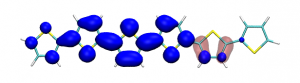
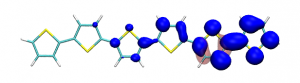
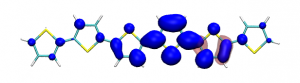
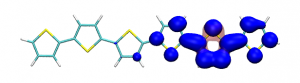
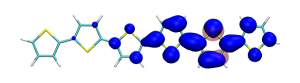
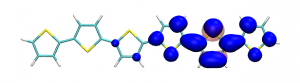
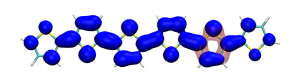
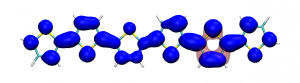
Splitting N2 is a challenging undertaking due to the strong triple bond holding the two nitrogen atoms together. Traditionally, this task is achieved via the Haber-Bosch process but recently interest has shifted to the possibilities of nitrogen splitting via homogeneous catalysis. Computations on the dinuclear transition metal complexes involved can be performed thanks to modern computational hardware and appropriate computer codes. However, the analysis of the excited states involved can become a significant challenge due to the large number of states and difficulties in assigning their character unambiguously. To tackle this problem, we have recently devised a strategy for a detailed analysis of the electronic wavefunctions of the states contributing to the reactive process. The associated paper, led by S. Rupp and V. Krewald from TU Darmstadt, just appeared in the European Journal of Inorganic Chemistry: Multi‐tier electronic structure analysis of Sita’s Mo and W complexes capable of thermal or photochemical N2 splitting.