A new study led by J. Lachner from the Helmholtz-Zentrum Dresden describes a method for detecting 26Al via Ion-Laser Interaction Mass Spectrometry using a particle accelerator.
Quantum chemical calculations highlight the different energetics of 26MgO and 26AlO, which are separated with high specificity despite being isobars.
A new study led by C. Heshmatpour and J. Hauer from TU München studies exciton-exciton annihilation in a squaraine trimer. The experiment exploits 5th-order optical spectroscopy to study the evolution of the trimer after two-photon excitation into its bi-exciton state. Quantum chemistry computations performed by M. Menger, now located at Groningen, provide the required parameters to model the experimental signals within a Frenkel exciton model. The associated article Annihilation Dynamics of Molecular Excitons Measured at a Single Perturbative Excitation Energy just appeared in J. Phys. Chem. Lett.
Quantum chemical computations were used to aid in the assignment of the structures produced and characterised via infrared multiple photon dissociation spectroscopy. An interactive model showing the relevant molecular vibrations can be found here.
Characterising excited states in transition metal complexes by looking at pictures of orbitals can be a tedious task. Even more, it is hard to eliminate personal in the process and produce quantitative results. In a study led by Pedro Sánchez-Murcia from the University of Vienna, we have taken a closer look at this problem in the case of various substituted complexes deriving from the archetype Ru(bpy)3 with the aim of quantifying how different substituents influence the localisation of the excited electron. The result is presented in the article “Orbital-free photophysical descriptors to predict directional excitations in metal-based photosensitizers,” which just appeared in Chemical Science.
Columbus is a collection of programs for high-level ab initio electronic structure computations. Through the use of multireference methods even highly challenging systems such as excited states and open-shell molecules are accessible. The availability of gradients and nonadiabatic coupling vectors allows for photodynamics simulations describing ultrafast internal conversion processes. The capabilities of Columbus have been showcased in a recent paper: The generality of the GUGA MRCI approach in COLUMBUS for treating complex quantum chemistry that just appeared in J. Chem. Phys. as part of a themed collection Electronic Structure Software.
A new release of the programm package, available to registered users, has been made available on the distribution page.
Singlet fission is a promising approach to photovoltaics where one pair of singlet charge carriers is converted intto two pairs of triplet charge carriers. Molecules undergoing singlet fission require a specific energetic condition: the first triplet state has to be half the energy of the first singlet state. In a study, lead by Max Pinheiro from Sao Paulo, Brazil, we investigated how this energetic condition can be achieved by substituting a tetracene molecule with B and N atoms in different positions. The resulting paper A systematic analysis of excitonic properties to seek optimal singlet fission: the BN-substitution patterns intetracene, which just appearred in J. Mat. Chem. C, shows the versatility of this approach in tuning the energies across a wide range. The results are rationalised by measuring unpaired electrons and compared to a more qualitative discussion in terms of bonding patterns and Clar sextets. We hope that the study will help by setting new accents in terms of design ideas for new chromophores.
For recent study presenting an alternative strategy of tuning energies for singlet fission, exploiting anti-aromaticity, see JACS 2019, 141, 13867.
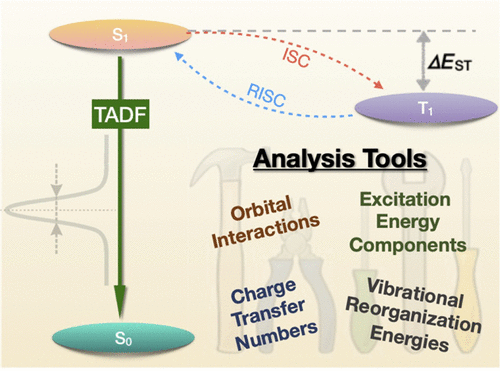
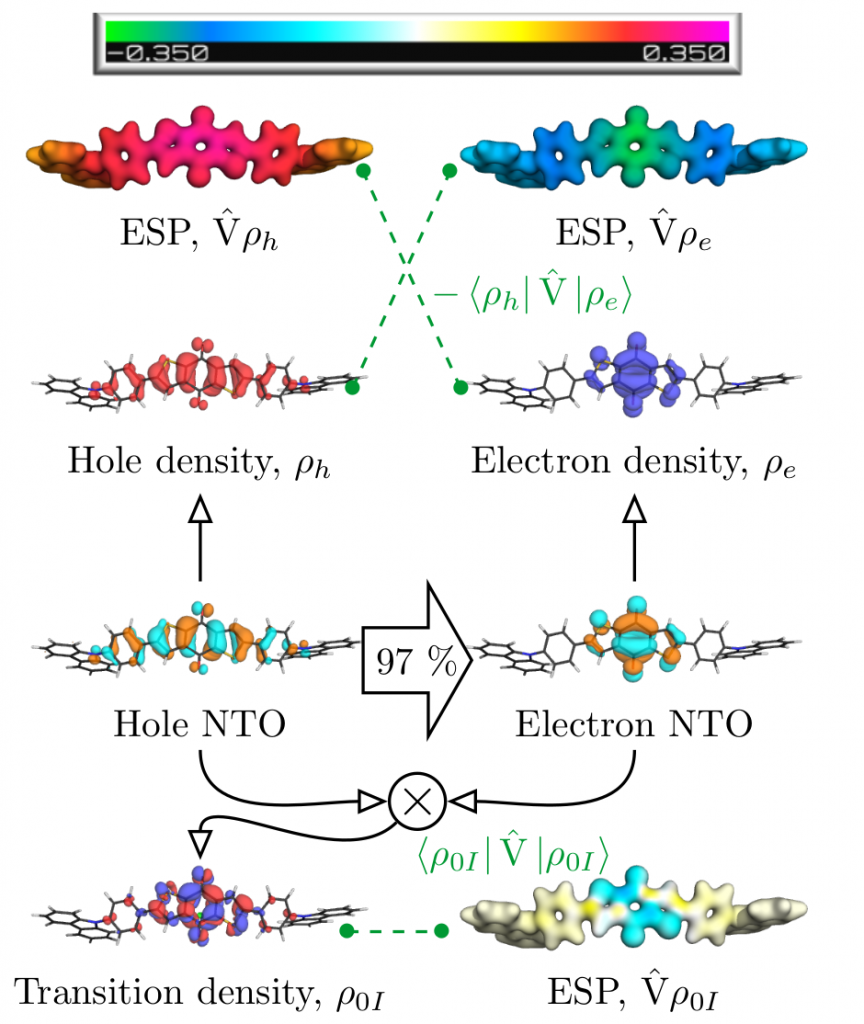
The molecular orbital (MO) picture has become the standard tool used by chemists to understand molecular electronic structure and particularly excited states. In our newest paper Toward an understanding of electronic excitation energies beyond the molecular orbital picture, which just appeared as a Perspective in PCCP, we asked whether we can go beyond the MO picture in a systematic way in our understanding of excited states. This endeavour ended up more involved than initially expected but we hope that it presents some interesting physics in a memorable and sufficiently intuitive way.
Below, we exemplify the tools developed to study a donor-acceptor-donor system initially developed in JACS, 2014, 136, 18070 and revisited in PCCP, 2019, 21, 10580. The HOMO of this system is on the carbazole donor whereas the LUMO is on the anthraquinone acceptor. The lowest excited state of the system (in gas phase) is not reached via the HOMO-LUMO transition but it is a locally excited state on anthraquinone. Why?
Figure 1: Analysis of post-MO contributions of the first excited singlet state of a donor-acceptor-donor molecule.
The contributions affecting the energy of the first excited singlet state are shown in Figure 1, above. In the third row the natural transition orbitals (NTOs) are shown. These give rise to the density of the excitation hole (red) and the excited electron (blue), shown above. The Coulomb attraction between these two charge distributions now stabilises the excited state. This Coulomb attraction is represented by the electrostatic potentials (ESPs) induced by the charge distributions. The important observation is that these ESPs are non-uniform with stronger contributions in the centre. Thus, the locally excited state is stabilised and an energetic penalty is paid for charge transfer. At the bottom of Figure 1, the transition density is shown, which leads to a repulsive exchange interaction destabilising local singlet states. In the present case the Coulomb attraction dominates and the lowest state is fairly local in nature.
Privacy & Cookies: This site uses cookies. By continuing to use this website, you agree to their use.
To find out more, including how to control cookies, see here:
Cookie Policy