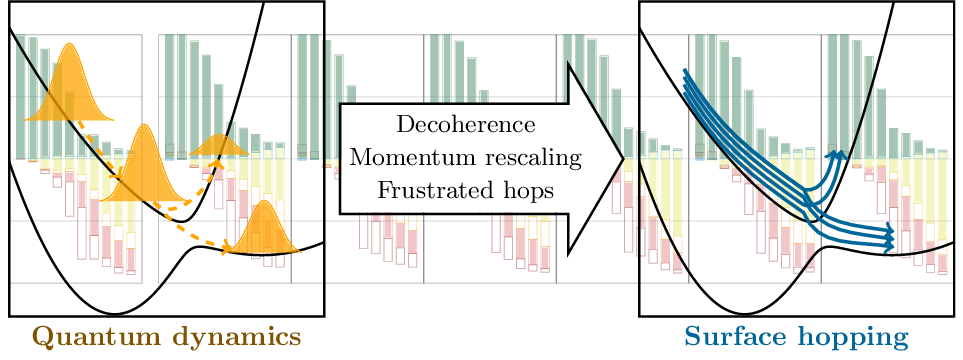
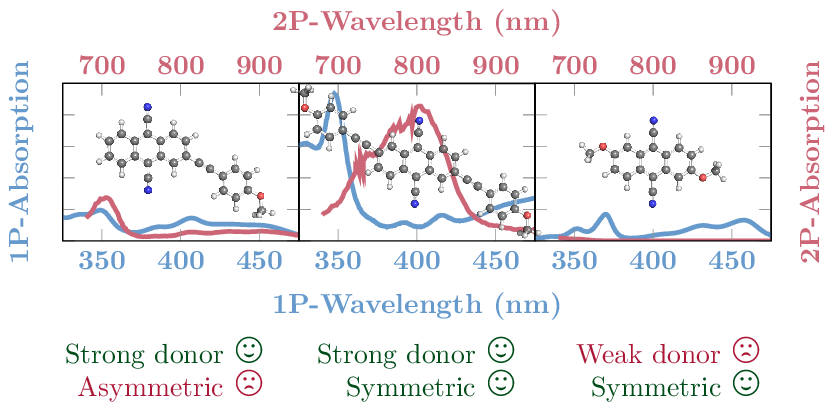
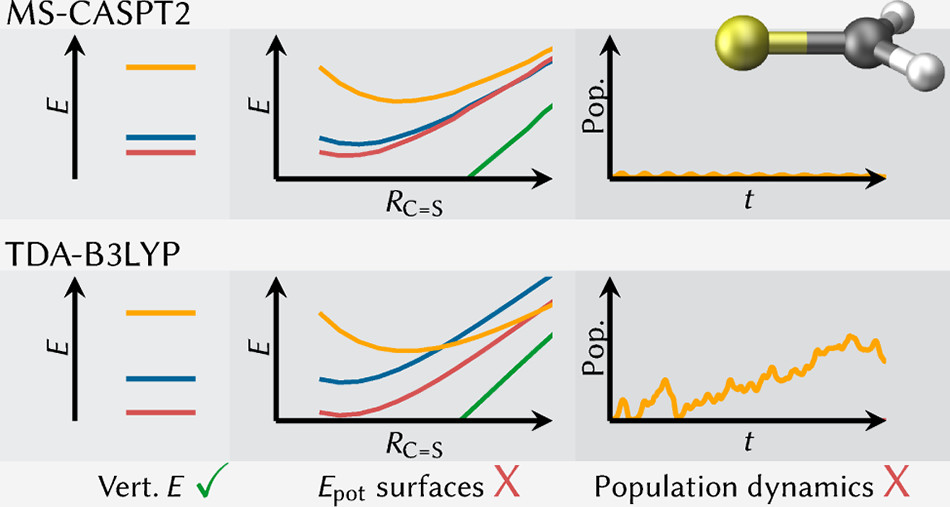
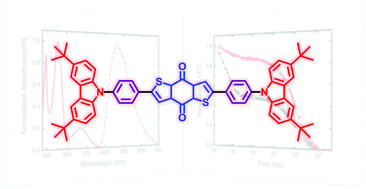
Tomorrow, Felix will give a talk at the Zernike Institute for Advanced Materials, Groningen. The talk is entitled: Understanding electronic excitation energies within and beyond the molecular orbital picture. It discusses how we can understand excited-state energies beyond simply looking at orbitals and their energies.
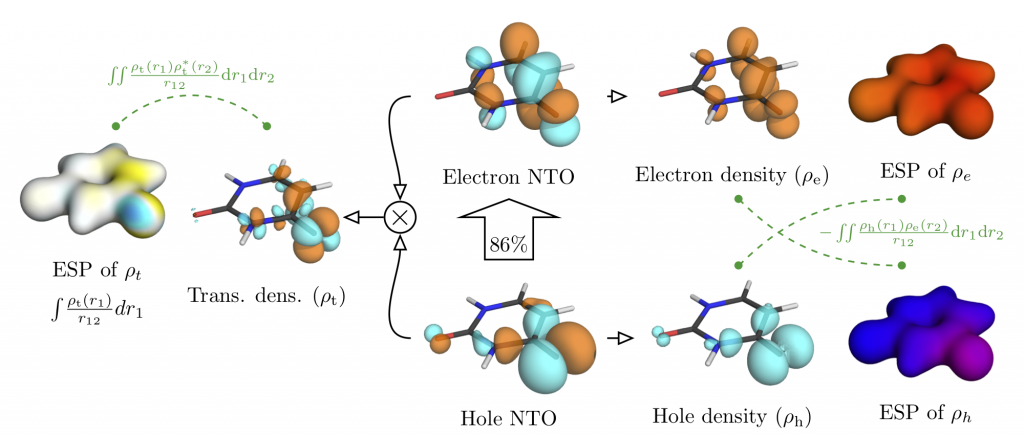
You can download the talk here: