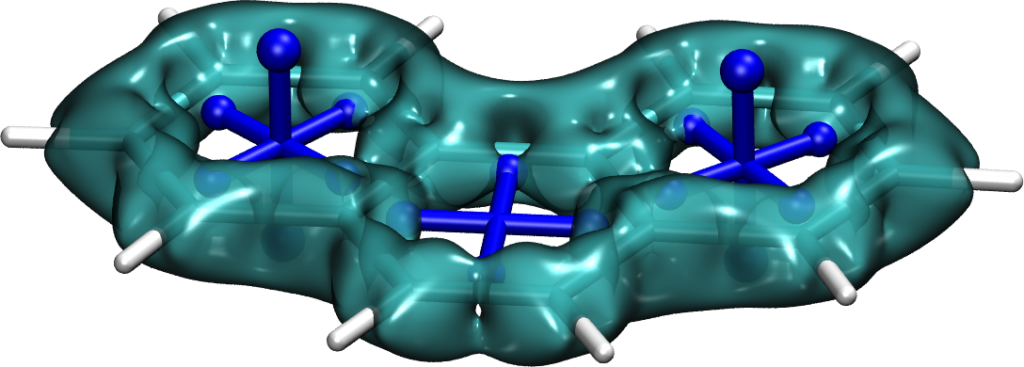
Can you sort these molecules according to increasing triplet excitation energies?
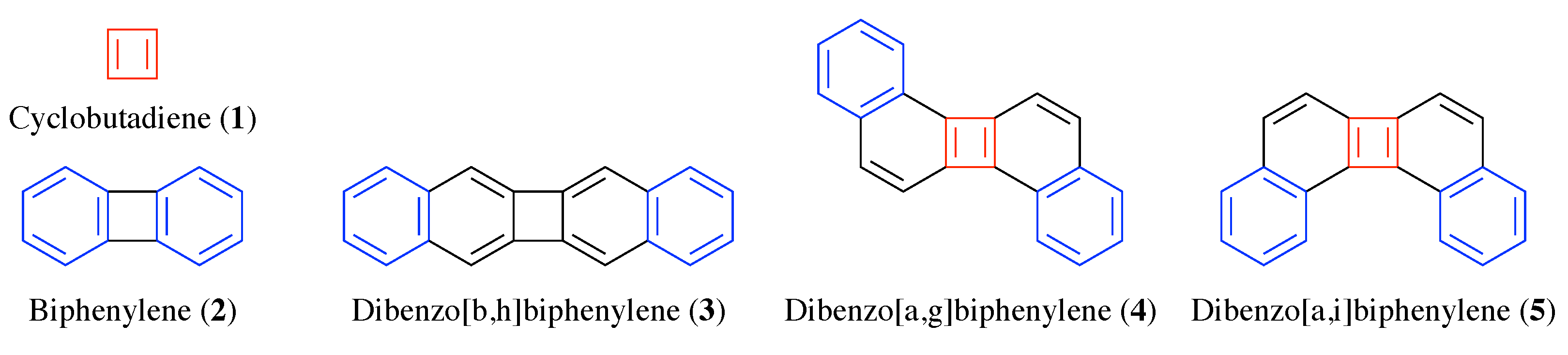
Some basic considerations might suggest that energies go down as the size of the molecule increases. But this is incorrect. The decisive feature of these molecules is their ground-state antiaromaticity along with their potential for excited-state Baird aromaticity. Triplet excitation energies increase sharply going from 1 (0.1 eV) via 2 (1.9 eV) to 3 (2.6 eV). This can be understood in the sense that antiaromaticity is blurred as the molecule becomes larger.
More strikingly, when going from 3 to 4 or 5, the energy drops again dramatically down to 1.0 eV. This effect is explained following Ayub et al. by the simple fact that these molecule possess resonance structures with simultaneous quartets and sextets.
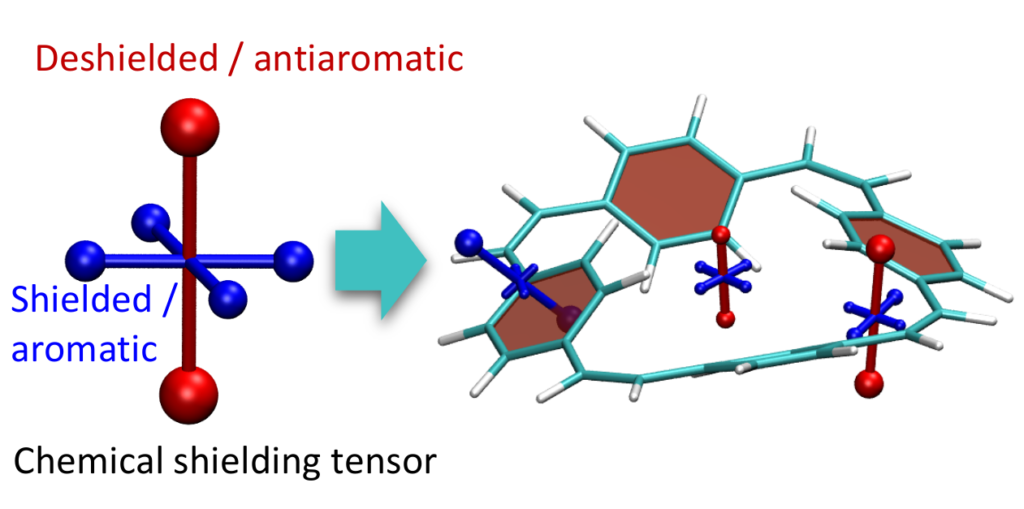
In a recent paper, Exploitation of Baird Aromaticity and Clar’s Rule for Tuning the Triplet Energies of Polycyclic Aromatic Hydrocarbons, we investigate these phenomena in detail using a recently developed method for the visualisation of chemical shielding tensors (VIST) along with an analysis of natural transition orbitals. In addition, a model for rationalising the dia- and paramagnetic shielding effects observed in (anti)aromatic systems is presented.
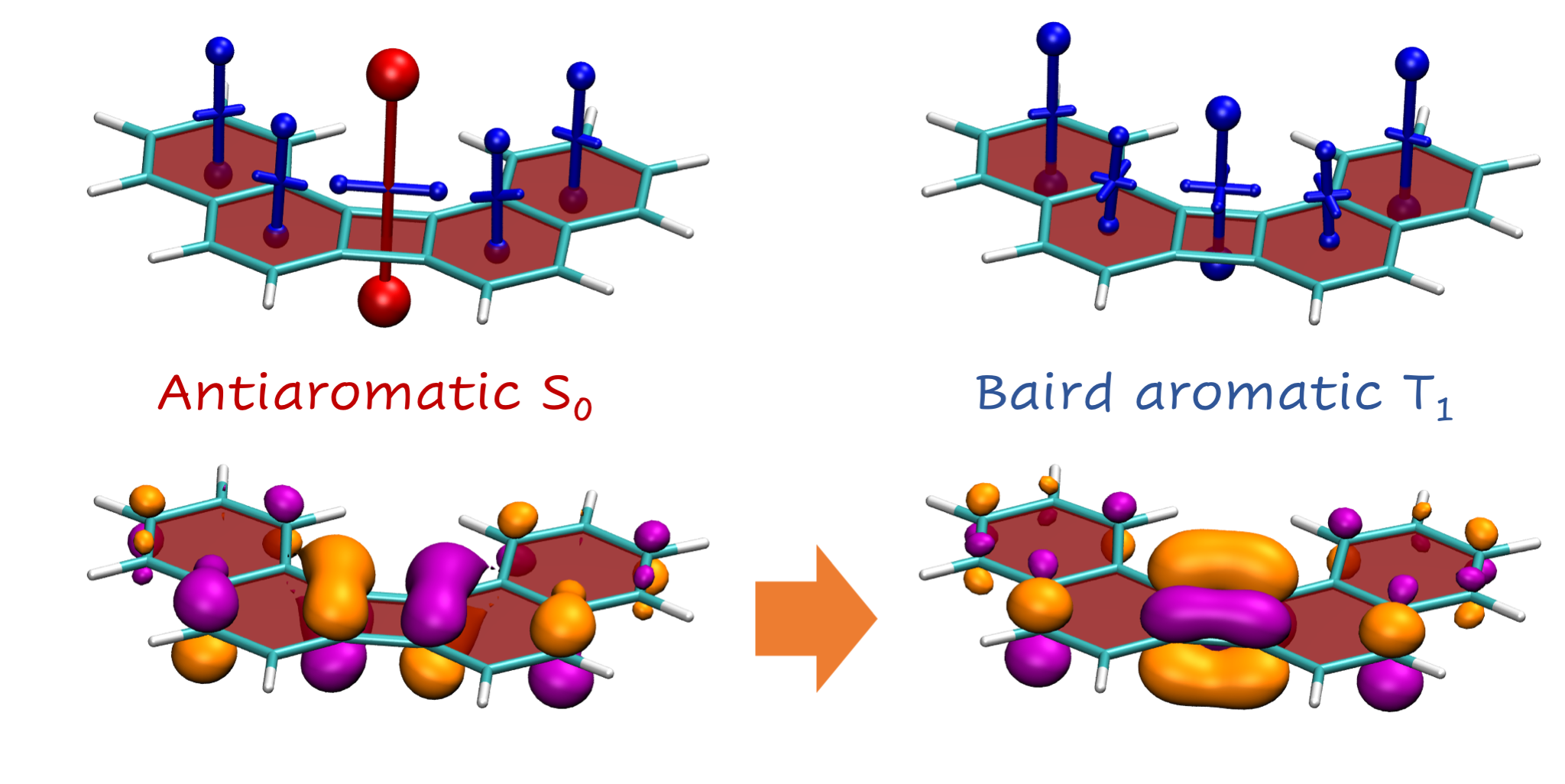
See also TCA, 2020, 139, 113 on a discussion of related bipenylene derivatives.