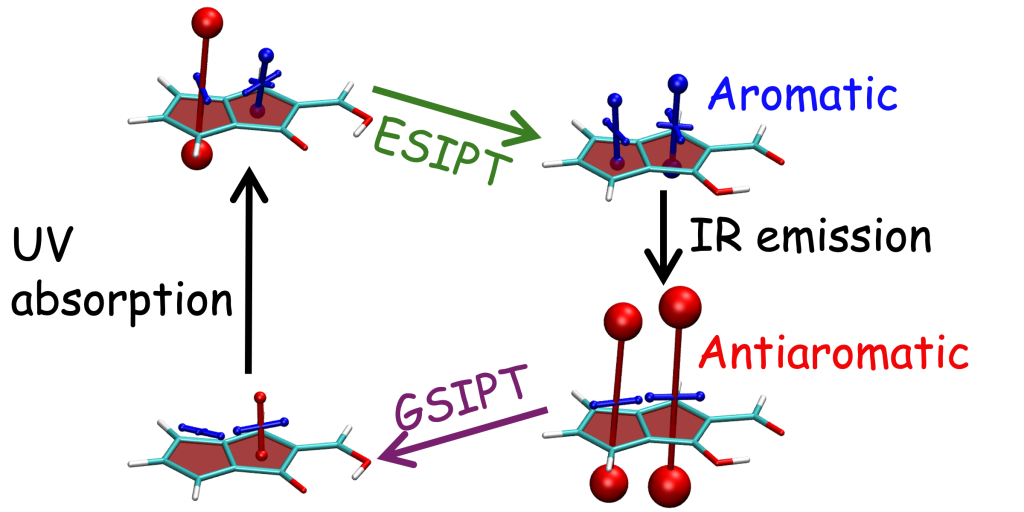
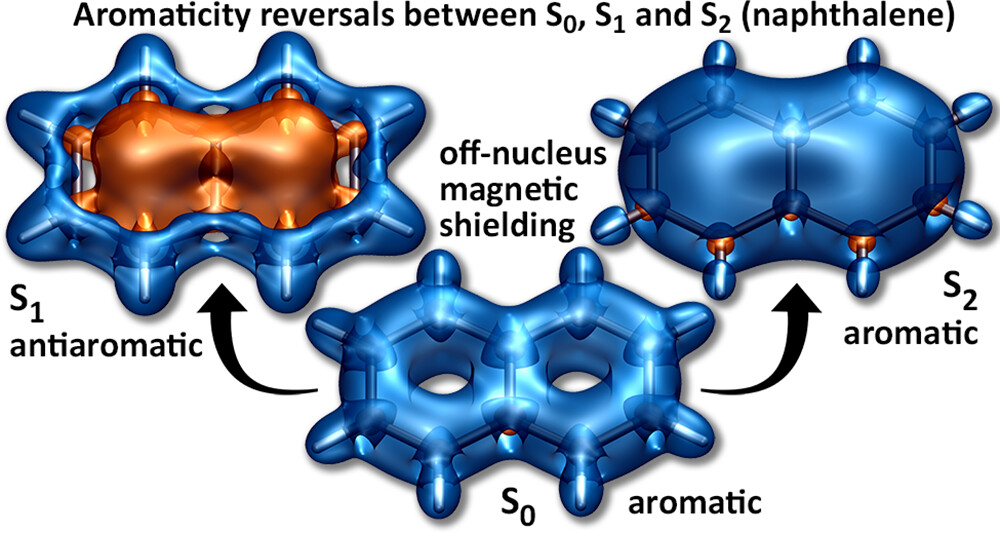
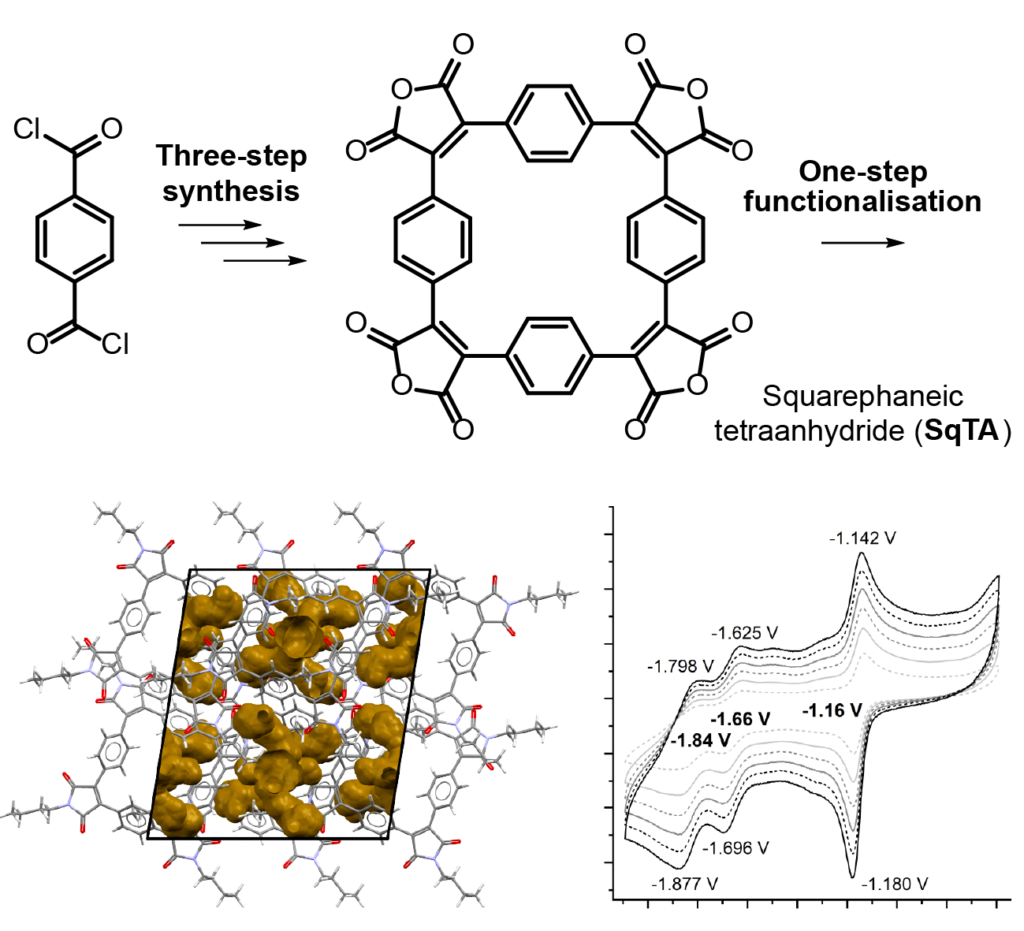
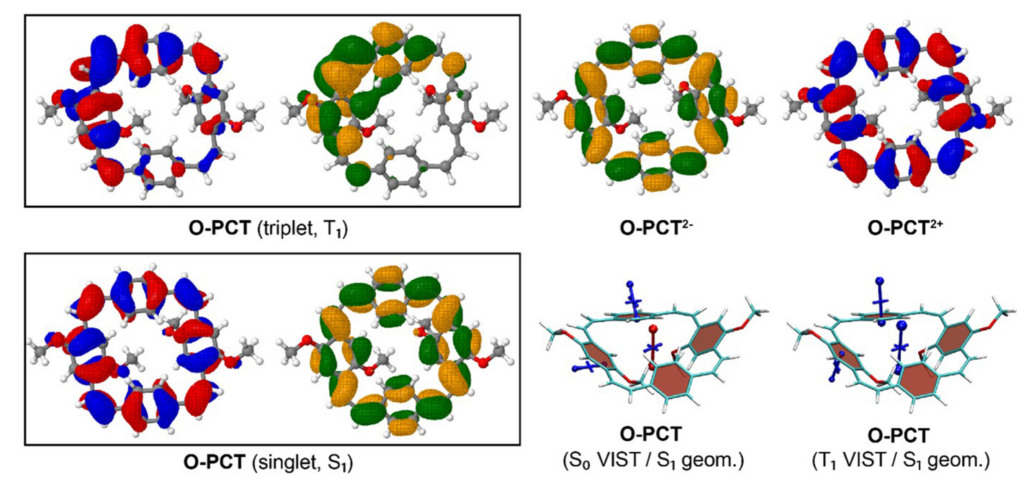
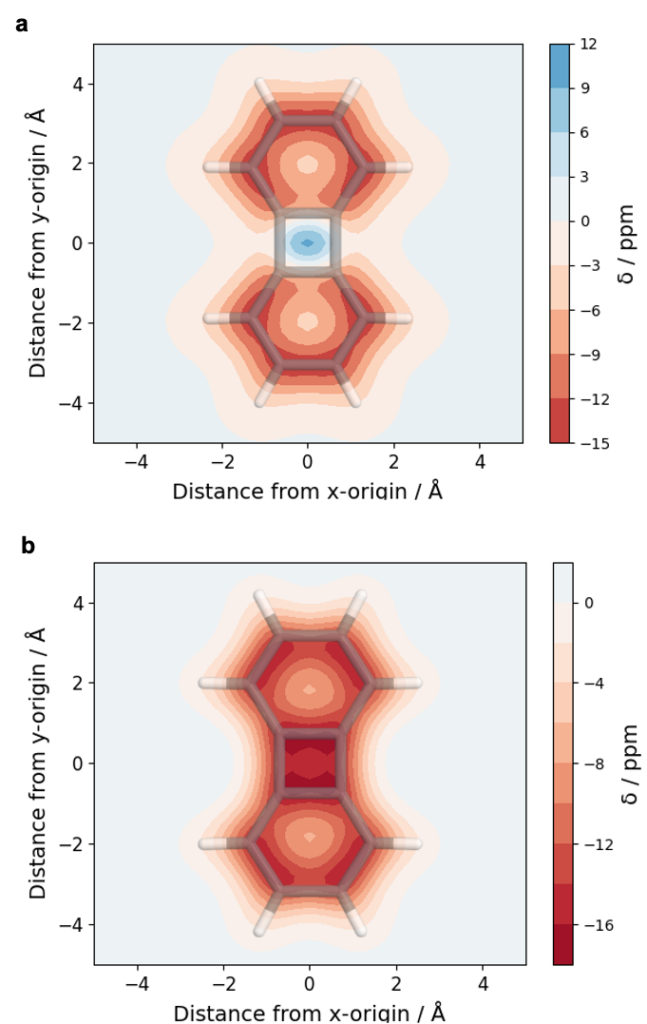
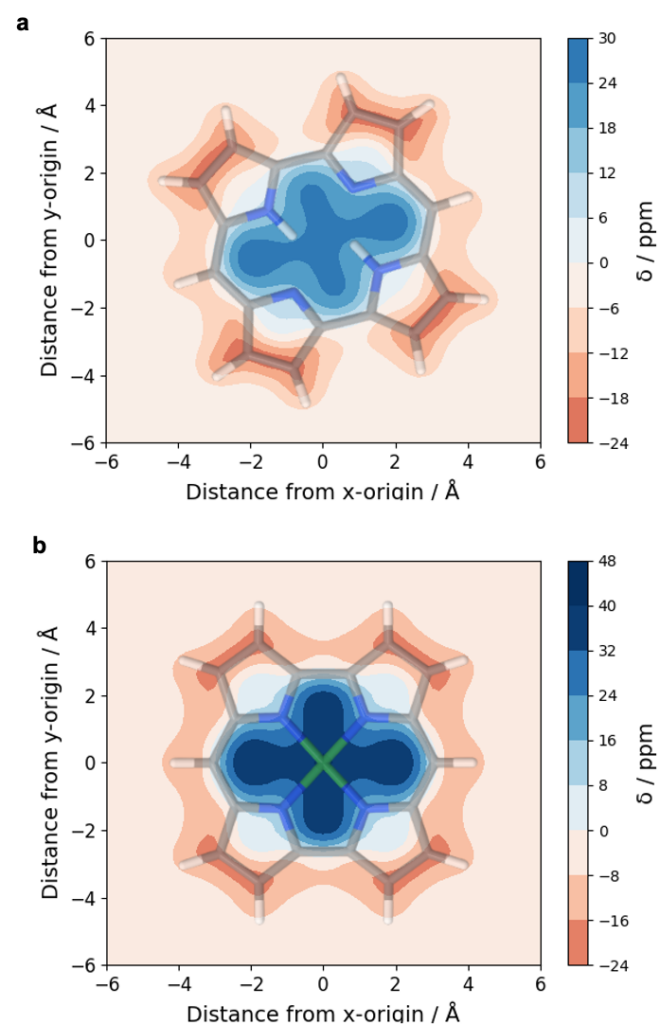
It is by now fairly well understood how chromophore properties are affected by push/pull substituents and their degree of planarity. But how can we rationalise variations in the properties of planar chromophores that do not possess charge transfer character?
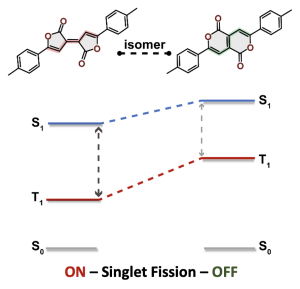
We were interested in understanding the apparent differences between these two isomeric molecules (called the Pechmann dyes).
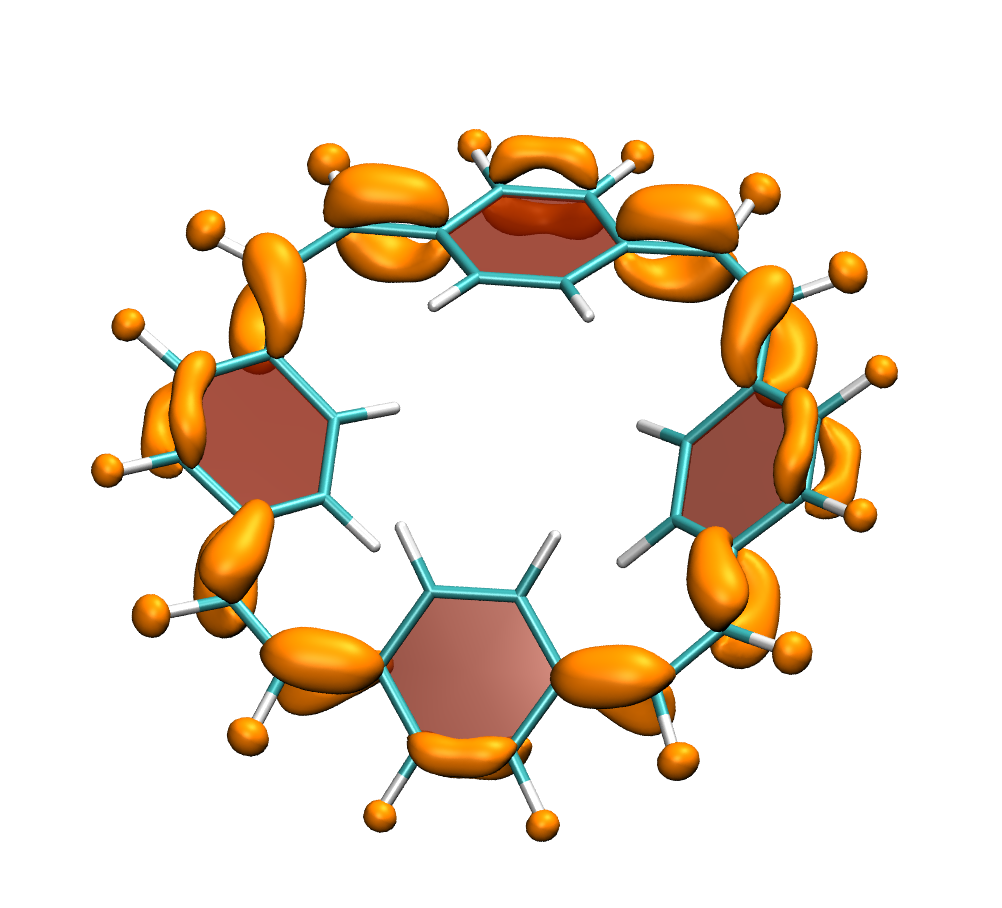
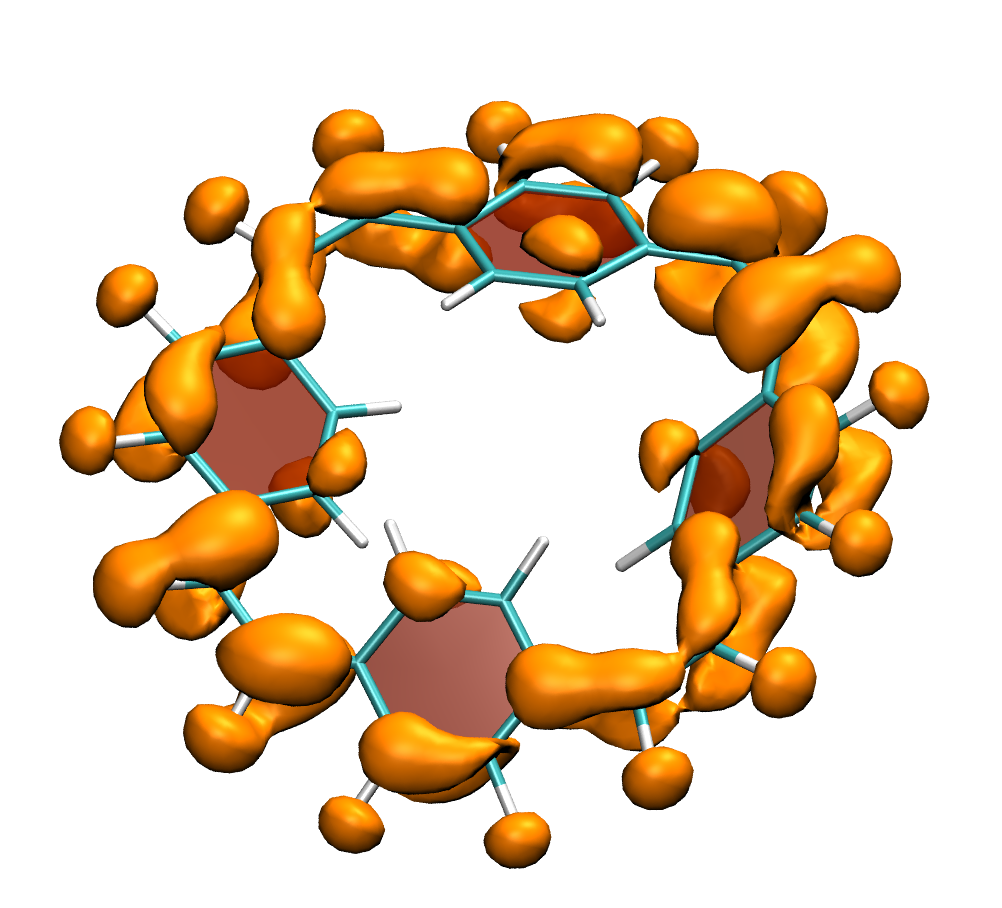
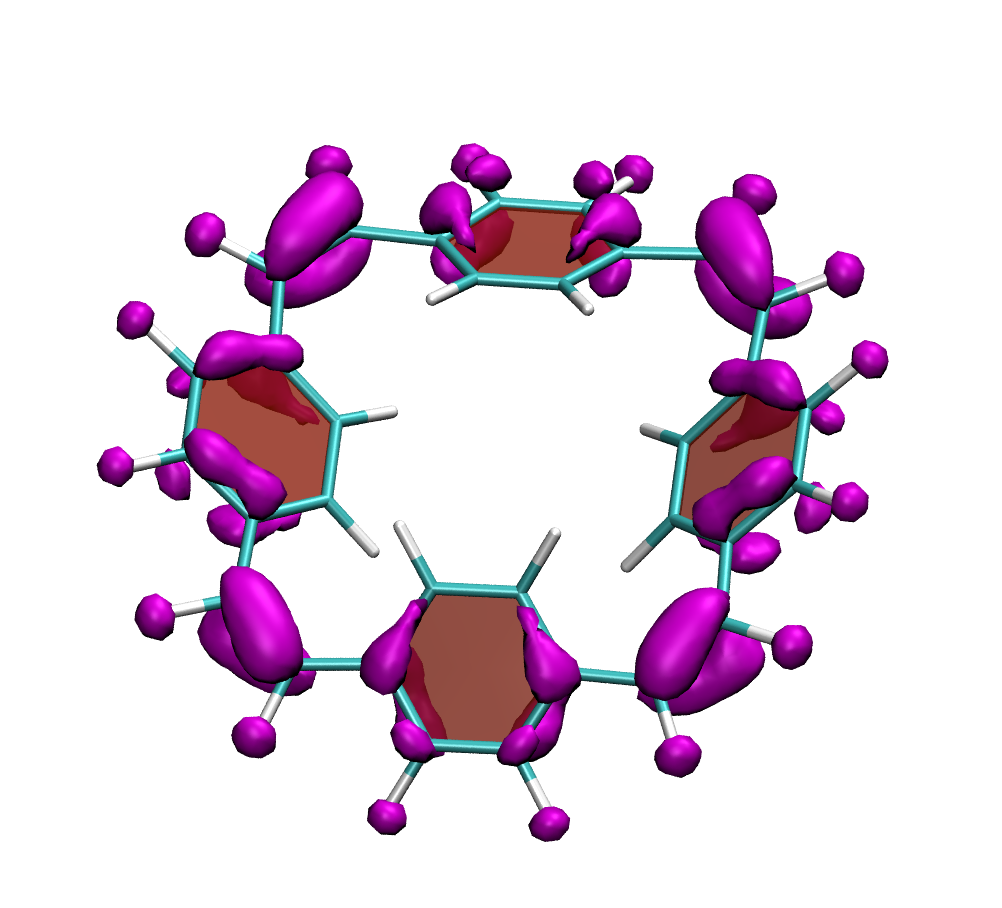
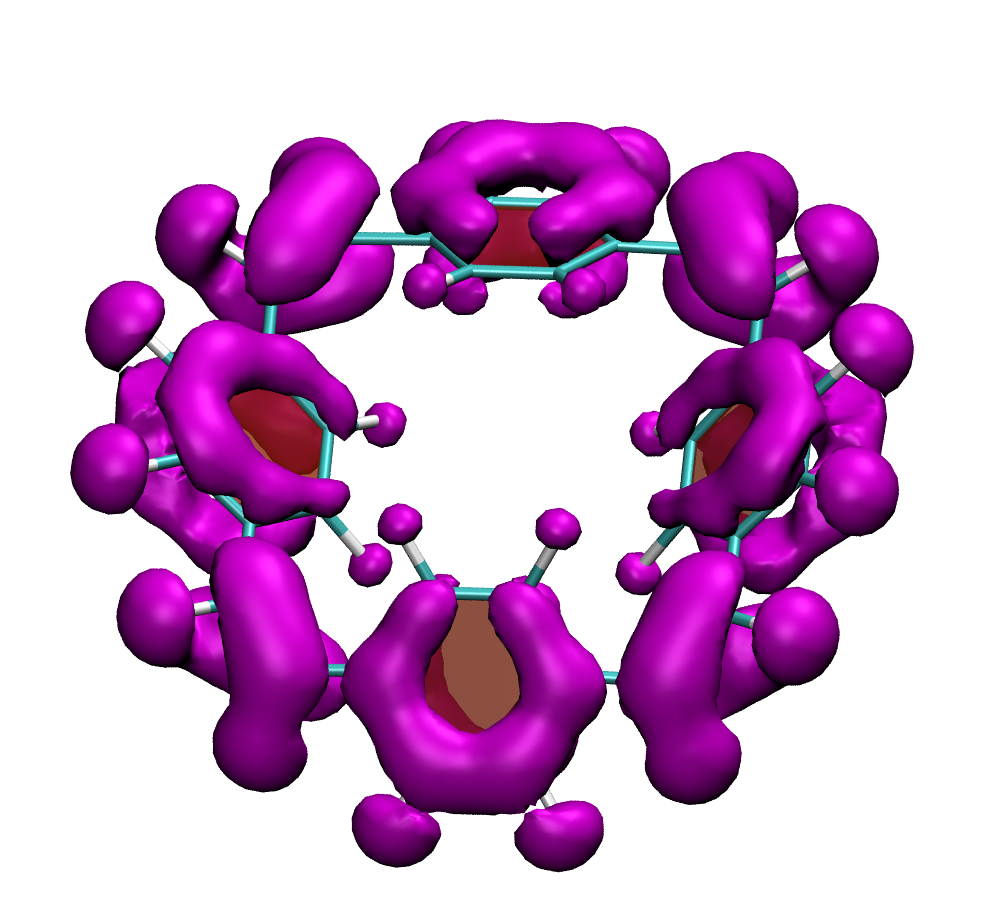
Why is the T1 of PM5 (shown to the left) so much lower than the one of PM6 (shown to the right) making PM5 a powerful candidate for a singlet fission material whereas the S1/T1 gap of PM6 is too small for this purpose?
The answer is discussed in the new paper “Singlet Fission in Pechmann Dyes: Planar Chromophore Design and Understanding” that just appeared in JACS.
The overall lower excitation energies of PM5 vs PM6 can be understood by the fact that the S1 and T1 of the former are stabilised via excited-state aromaticity whereas the S0 of the latter profits from ground-state aromaticity.
The reason for the lower S1/T1 gap in PM5 is more subtle but also more fascinating pointing to a whole new way of viewing excitation energies. We were able to highlight the effect of the double bond conformation in influencing the exchange integral between the excited electron and hole, which ultimately leads to the variations in S1/T1 gap.