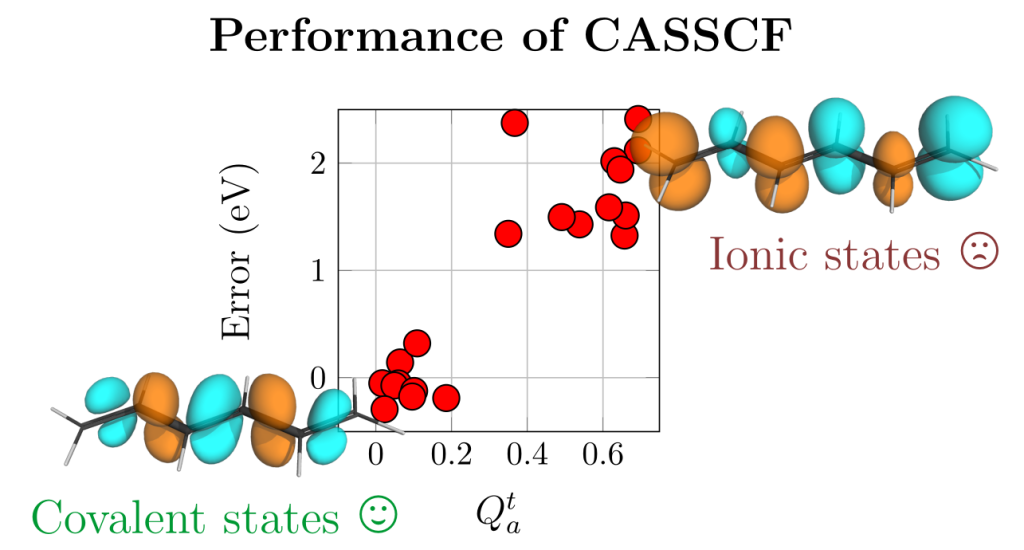
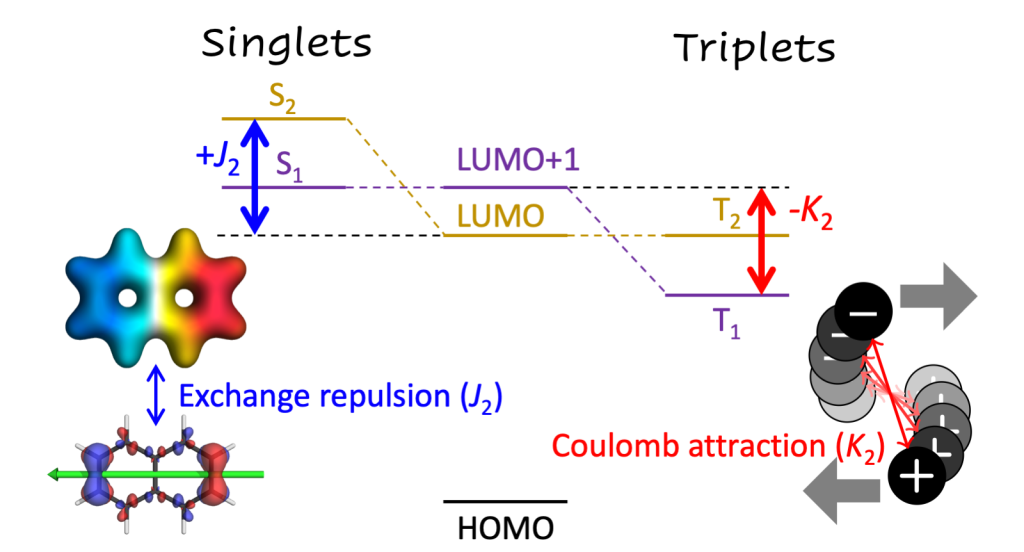
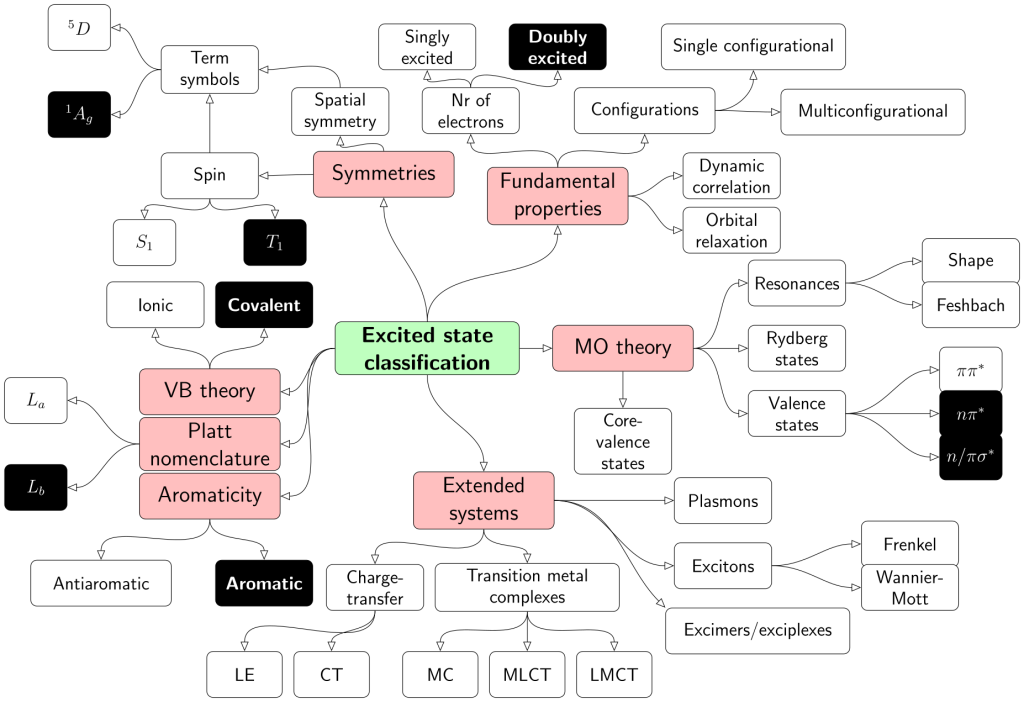
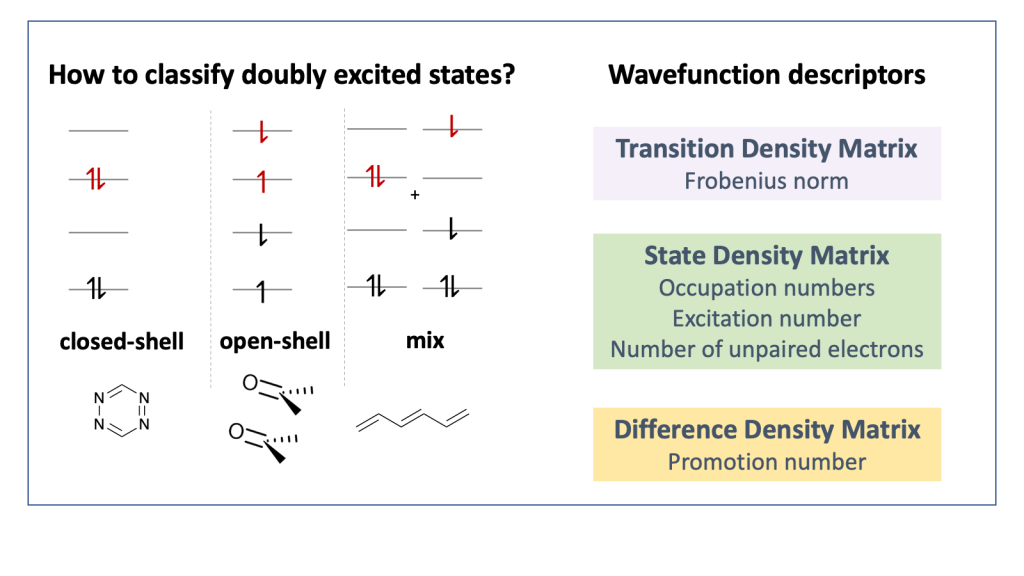
Computations on diradicals are not only difficult in terms of choosing an appropriate electronic structure method but in many cases it is also quite challenging to make sense of the results obtained. To tackle this problem we developed a detailed characterisation scheme for the excited states of diradicals in our new paper Classification and quantitative characterisation of the excited states of π-conjugated diradicals that just appear in Faraday Discussions.

Our paper builds on earlier work by Salem et al. and Stuyver et al. in terms of formally characterising the states as diradical and zwitterionic within a two-orbital two-electron model (TOTEM). On top of this, we have provided practical protocols for recognising these states in realistic computations. Using our tools in the case of the para-quinodimethane molecule as a model system, we show that it is possible to identify the states arising from the TOTEM but also that the π-conjugated bridge plays a crucial role producing more complicated states than one would have expected from the simple TOTEM.