Kasha’s rule states that fluorescence generally occurs from the lowest excited singlet state (S1). Exceptions to this rule are usually associated with a metastable S2 state that is separated from S1 not allowing for interconversion. In a recent article we outlined a different mechanism for non-Kasha fluorescence: If S1 and S2 are very close in energy, then S2 is populated in a dynamic equilibrium following Boltzmann statistics. This effect is particularly pronounced if there is a large amount of vibrational excess energy following excitation into a high-energy absorption peak. The full story, “Non-Kasha fluorescence of pyrene emerges from a dynamic equilibrium between excited states” was just published in J. Chem. Phys.
Paper
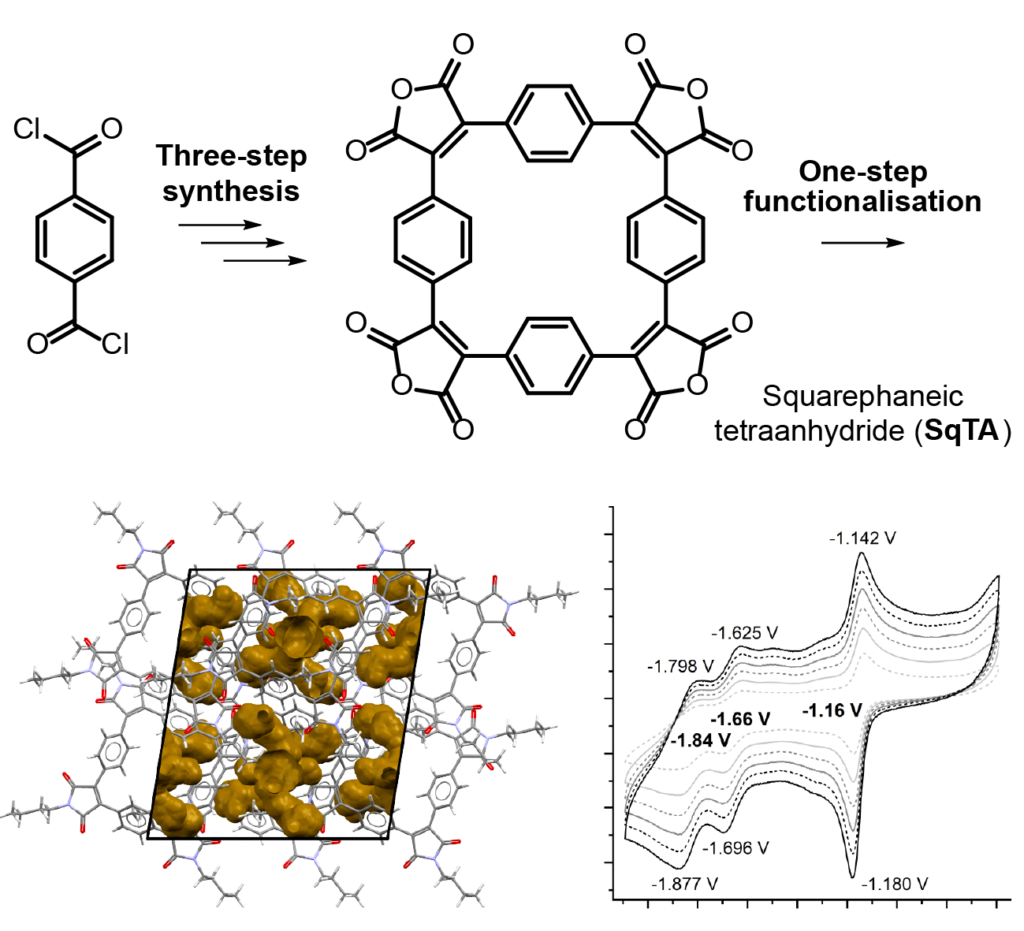
Squarephaneic Tetraanhydride
Our new paper “Squarephaneic Tetraanhydride: A Conjugated Square-Shaped Cyclophane for the Synthesis of Porous Organic Materials” was just published in Angewandte Chemie. This paper, led by Florian Glöcklhofer from Imperial college, explores the redox chemistry, porosity, and chemical derivatives of a newly developed tetraanhyride based on a macrocycle with a formally antiaromatic ground state.
Hydrogen-bonded Organic Framework
Our new paper “Hierarchical Assembly of a Micro-and Macroporous Hydrogen-Bonded Organic Framework with Tailored Single-Crystal Size,” led by Antonio Fernandez at Loughborough, just appeared in Angewandte Chemie.
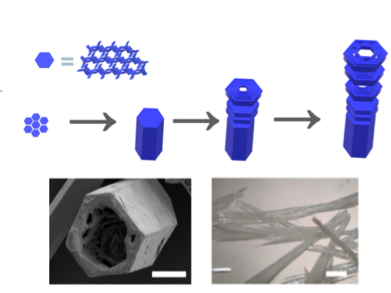
The work shows how a highly porous framework can be constructed by using different types of intermolecular interactions.
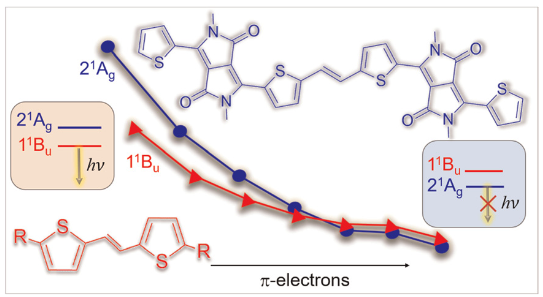
Diketopyrrolopyrroles
Our new paper Using diketopyrrolopyrroles to stabilize double excitation and control internal conversion, led by Mariana do Casal and Mario Barbatti from Aix Marseille University, just appeared in PCCP. This work highlights how double excitation character can support internal conversion. Wavefunction analysis using TheoDORE sheds light into the wavefunctions involved.
Eu(III) complexes
Our new paper “Impact of Varying the Phenylboronic Acid Position in Macrocyclic Eu(III) Complexes on the Recognition of Adenosine Monophosphate“, led by S. E. Bodman and S. J. Butler from Loughborough, just appeared in Organic Chemistry Frontiers. The paper is the second in a series studying the anion sensing properties of Eu(III) complexes with phenylboronic acids.
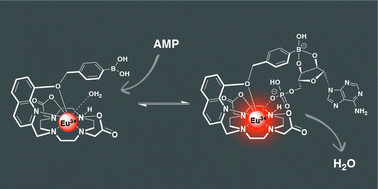
Aside from reporting the synthesis and anion binding, the paper presents new strategies for the computational analysis of such complexes. Aside from modelling the geometries by density functional theory, high-level multireference methods in OpenMolcas were applied to study the luminescence properties. These first principles computations offer a promising approach to access the emission spectra of lanthanide complexes, aiding the design of responsive lanthanide probes with specific photophysical properties
Substituted macrocycles
A recent study, lead by Florian Glöcklhofer from Imperial College London, explores the effect of methoxy and thiomethyl subtitutions on a formally antiaromatic macrocycle. The corresponding paper “[2.2.2.2]Paracyclophanetetraenes (PCTs): cyclic structural analogues of poly(p‑phenylene vinylene)s (PPVs)” is available via Open Research Europe, 1, 111, 2012.
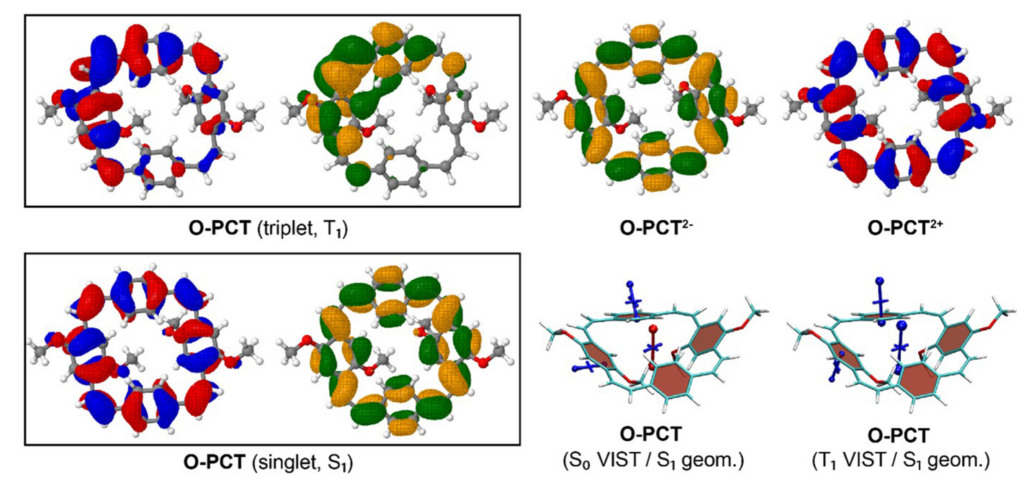
The above figure compares the orbitals and aromaticity descriptors for different charge and spin states. Importantly, the symmetry is broken in the T1 state, inhibiting Baird aromaticity. By comparison, the symmetry is retained for the neutral singlet, dianion, and dication states all of which exhibit aromaticity.
Reversible P-P bond cleavage
Recent work out of the group of Martin Smith at Loughborough University presents the possibility of cleaving a P-P bond at an iridium(III) metal centre by adding an AuCl unit.
Computations elucidate the underlying energetics and rationalise the results using the natural bond orbitals (NBO) approach.

You can find the full text as an Advance Article in Chem. Commun.: Reversible P–P bond cleavage at an iridium(III) metal centre.
Luminescent lanthanide probes
Recent research led by Samantha Bodman and Steve Butler from Loughborough University presents a luminescent lanthanide probe with selective affinity for adenosine monophosphate (AMP), able to differentiate AMP from the more highly charged analogues ADP and ATP.
Density functional theory computations shed insight onto the binding modes involved.
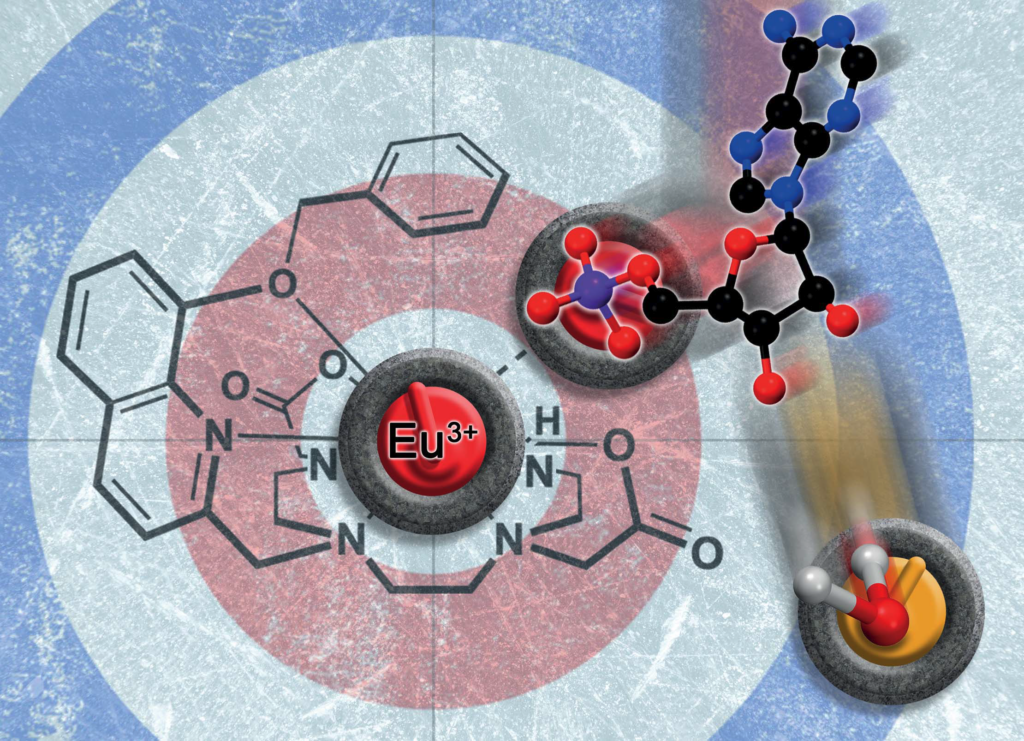
You can find the full article at Chem. Sci. 2022, 13, 3386.
libwfa: Wavefunction analysis tools
Our new paper “libwfa: Wavefunction analysis tools for excited and open-shell electronic states” was just published in WiRES Comp. Mol. Sci. The libwfa library provides a variety of visual and qunantitative analysis tools to post-process excited-state computations performed within Q-Chem and OpenMolcas.
You can find the libwfa functionality in the Q-Chem documentation under General Excited-State Analysis. To activate libwfa in Q-Chem, use
state_analysis = true
In OpenMolcas use the WFA module, activated via
&WFA
Delayed fluorescence
Patrick’s first paper as first author just appeared in PCCP: The role of excited-state character, structural relaxation, and symmetry breaking in enabling delayed fluorescence activity in push-pull chromophores. Well done Patrick!
We were interested in understanding the difference in thermally activated delayed fluorescence (TADF) between two closely related donor-acceptor-donor systems using either an anthraquinone and benzodithiophenedione acceptor units, respectively. The first one was known to be an effective TADF emitter [JACS 2014, 136, 18070] whereas the second one had significantly lower quantum yield for TADF [PCCP 2019, 21, 10580].
Rather than just presenting energies, it was the purpose of this paper to shed detailed insight into the wavefunctions involved. Notable differences in the wavefunctions and charge-transfer character were found between the two molecules. Even more striking differences existed between different computational methods.
After evaluating electronic structure methods, we presented geometry optimisations in solution, highlighting the importance of symmetry breaking for producing an emissive lowest singlet state. The role of different solvation models was discussed as well.