We are organising a new summer school for PhD students and early postdocs:
Quantum Chemistry of Excited States (QCES-2024)
The school will take place from 25-29 November 2024. Please have a look and see if you are interested.
We are organising a new summer school for PhD students and early postdocs:
Quantum Chemistry of Excited States (QCES-2024)
The school will take place from 25-29 November 2024. Please have a look and see if you are interested.
Applications are invited for a postdoctoral research associate position in computational chemistry at Loughborough University. This is a DSTL funded position to work with Dr Kenny Jolley and Dr Felix Plasser (Chemistry, Loughborough University) on predicting the crystal structure, dynamics and physical properties of energetic materials.
The successful applicant will conduct molecular dynamics (MD) and accelerated MD simulations on energetic compounds. Bulk physical properties, stability to shock and heat, and reaction pathways will be modelled and compared to experimental data. In a later stage, we will perform time-resolved simulations of explosion processes.
This position is ideally suited for an ambitious early career researcher with a background in computational chemistry and materials modelling. The successful candidate will be highly motivated with a strong research track record and a desire to pursue multidisciplinary research.
Feel free to contact me or Dr Kenny Jolley for informal equiries.
Closing date for applications is 24/11/2023, please follow this link for further info.
An interview with Felix was just published by EuChemS magazine. You can have a look here.
A new funded PhD position is available in the group: Computational design of functional molecular materials.
This is a flexible position allowing you to apply state-of-the-art quantum chemistry along with sophisticated analysis methods. The goal is to develop new rules for designing functional materials going beyond the frontier orbital picture. The work will apply rules developed in PCCP, 22, 6058-6080 (2020) in connection with experimental partners.
This is a flexible studentship that can be adjusted to your needs and interests. Please apply by February 2023 if this interests you.
The COLUMBUS programme package – a collection of programs for high-level ab initio molecular electronic structure calculations – has been released open-source. Please find
Any contributions (from small bugfixes to major features) are welcome. You can find more information about contributing here.
Dylan has finished his MChem project entitled “Visualisation of Aromaticity and Antiaromaticity via the Computation of the Chemical Shielding on Multi-Dimensional Grids.” You can find his report here. The purpose of his project was to develop a convenient method for computing shielding tensors on a grid around a molecule. The developed code is available via github.
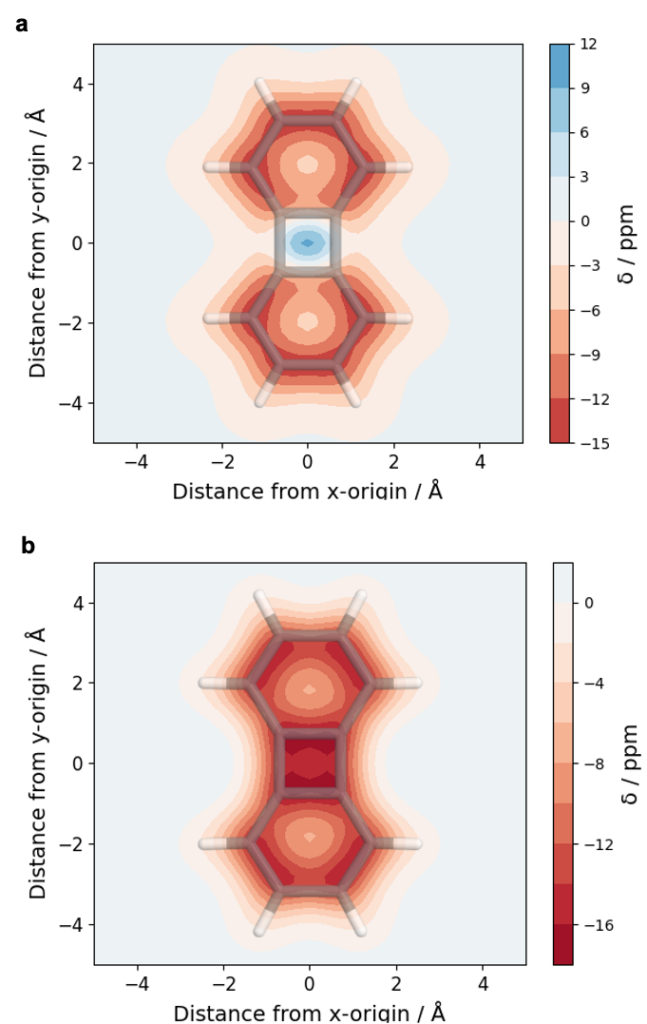
Below, an analysis of biphenylene is shown in the singlet (a) and triplet (b) state. For the singlet this representation highlights the aromaticity (red) of the benzene rings whereas the central 4-membered ring is found to be antiaromatic (blue). In the triplet (b), the whole molecule is found to be aromatic (red) according to Baird’s rule.
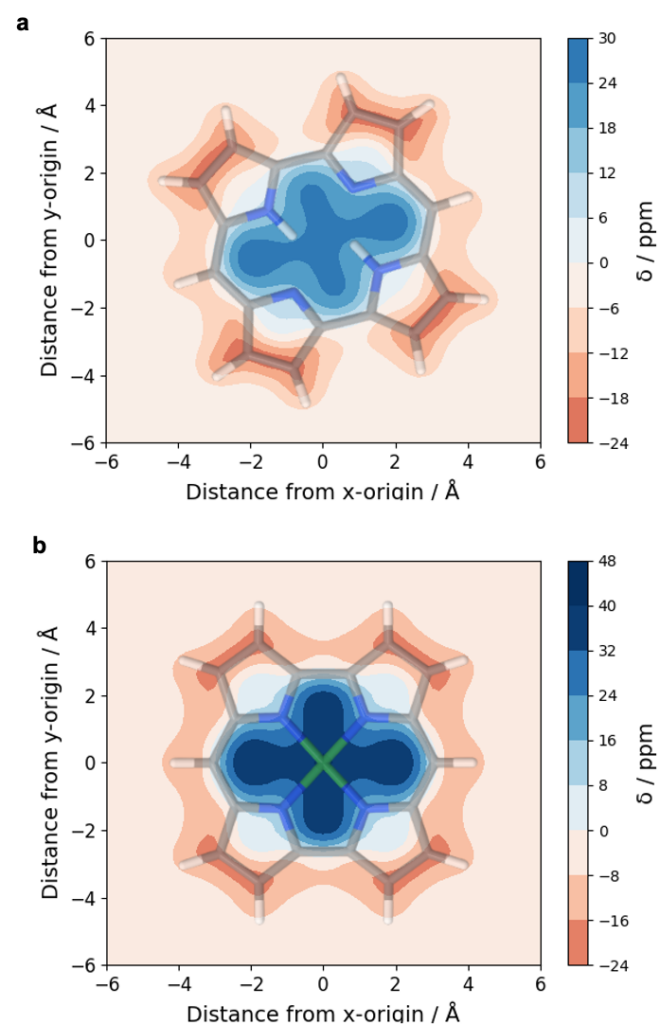
An analysis of norcorrole using either its doubly protonated form (a) or a nickel complex (b) highlights the antiaromaticity at the centre of this molecule whereas an aromatic pathway is found at the perimeter (see also [P. B. Karadakov, Org. Lett. 2020, 22, 8676]).
For more examples of how this type of analysis is used in the literature, see e.g., [Angew. Chemie – Int. Ed. 2020, 59, 19275] and [ChemPhysChem 2021, 22, 741].
We will host this year’s OpenMolcas developers’ e-meeting, taking place from 29 June to 2 July 2021. You can find more information here.
Dylan Morgan joined the group to work on his MChem project: “Visualisation of Aromaticity in Macrocycles”. The project is inspired by recent findings that anti-aromaticity in macrocycles provides a promosing route for the design of new battery anodes.
Aromaticity, despite its ubiquity in the discussions, is still surprisingly hard to visualise and quantify. We will endeavour to compare the different available techniques – nucleus-independent chemical shifts, current density plots, and the GIMIC method – with the goal of identifying the most promising ones and streamlining the workflows. In particular, we are interested in 1D, 2D, or even 3D scans of NICS values as inspired by a recent paper on excimers.
Welcome Dylan!
An update of the WFA module has been posted to OpenMolcas. This update integrates the fragment-based analysis that was previously only available via the TheoDORE code. In particular, it allows the automatic analysis of excited-state character in transition metal complexes [1, 2] with just a few added lines in the input file to OpenMolcas. This functionality is described here.
Thank you to Feng Chen from Loughborough University’s Research Software Engineering program for implementing the new code.
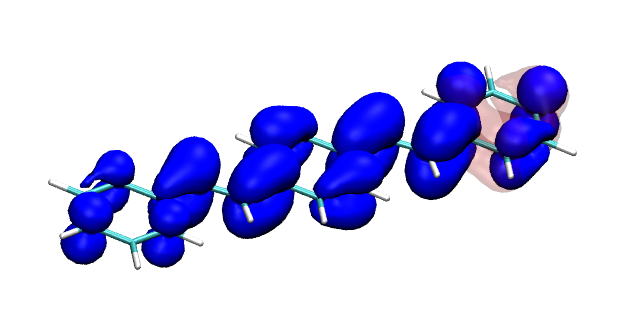
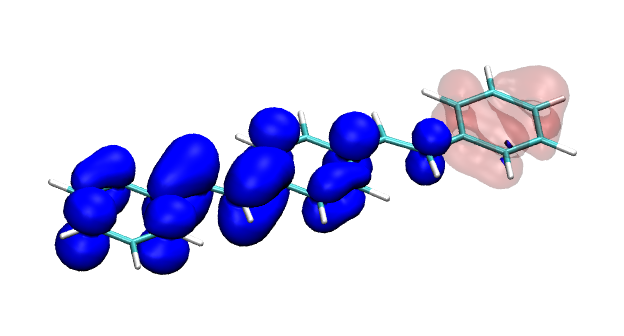
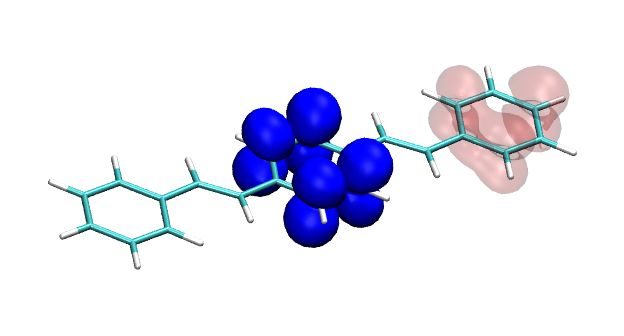
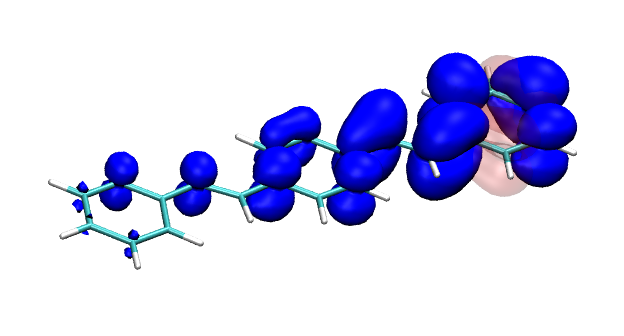
Version 2.0 of the TheoDORE wavefunction analysis package has been released, download below. The two main features of TheoDORE 2.0 are the computation of conditional electron densities and compatibility with python3.
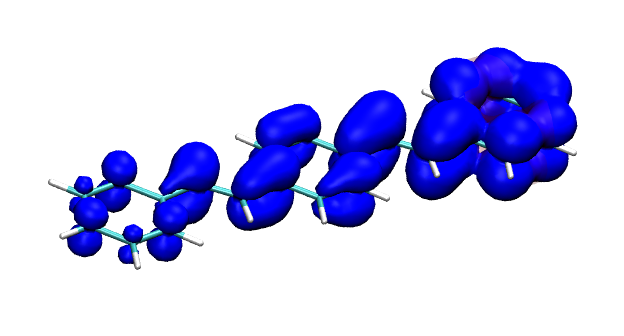
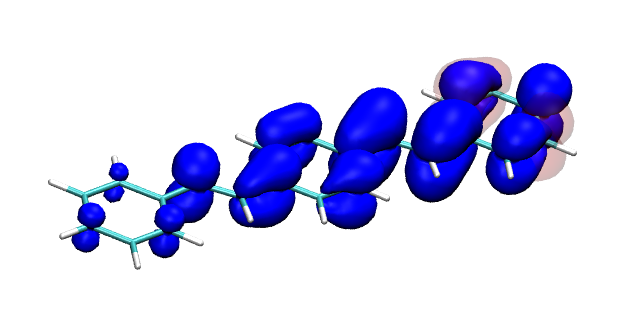
Conditional electron densities can be used for the visualisation of excited-state electron correlation, see ChemPhotoChem (2019). Below, the application of this method to a PPV oligomer is shown. Here, the probe hole (red) is always fixed on the terminal phenyl ring and the different shapes for the conditional electron density (blue) for the first six excited states is observed. One can see that for the different states the electron is either repelled, attracted or unaffected by the hole.