Columbus is a collection of programs for high-level ab initio electronic structure computations. Through the use of multireference methods even highly challenging systems such as excited states and open-shell molecules are accessible. The availability of gradients and nonadiabatic coupling vectors allows for photodynamics simulations describing ultrafast internal conversion processes. The capabilities of Columbus have been showcased in a recent paper: The generality of the GUGA MRCI approach in COLUMBUS for treating complex quantum chemistry that just appeared in J. Chem. Phys. as part of a themed collection Electronic Structure Software.
A new release of the programm package, available to registered users, has been made available on the distribution page.
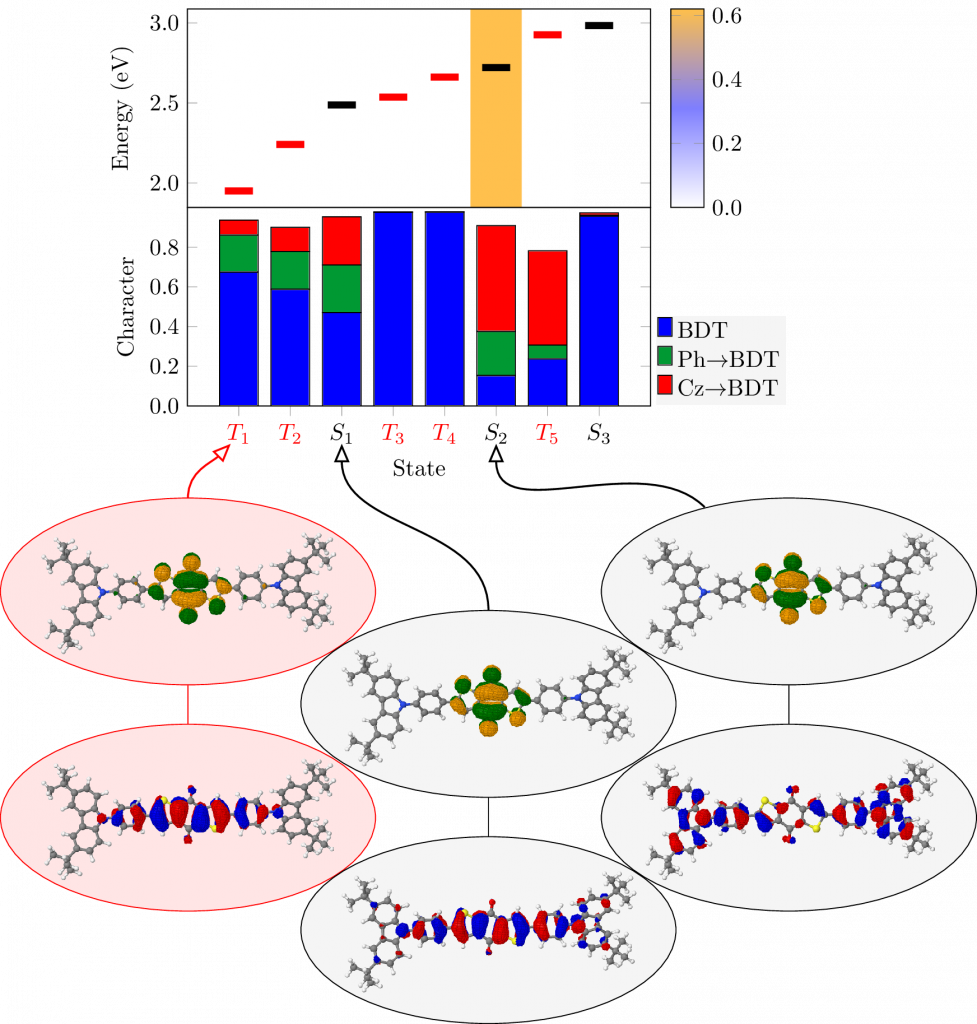
Singlet fission is a promising approach to photovoltaics where one pair of singlet charge carriers is converted intto two pairs of triplet charge carriers. Molecules undergoing singlet fission require a specific energetic condition: the first triplet state has to be half the energy of the first singlet state. In a study, lead by Max Pinheiro from Sao Paulo, Brazil, we investigated how this energetic condition can be achieved by substituting a tetracene molecule with B and N atoms in different positions. The resulting paper A systematic analysis of excitonic properties to seek optimal singlet fission: the BN-substitution patterns intetracene, which just appearred in J. Mat. Chem. C, shows the versatility of this approach in tuning the energies across a wide range. The results are rationalised by measuring unpaired electrons and compared to a more qualitative discussion in terms of bonding patterns and Clar sextets. We hope that the study will help by setting new accents in terms of design ideas for new chromophores.
For recent study presenting an alternative strategy of tuning energies for singlet fission, exploiting anti-aromaticity, see JACS 2019, 141, 13867.
The molecular orbital (MO) picture has become the standard tool used by chemists to understand molecular electronic structure and particularly excited states. In our newest paper Toward an understanding of electronic excitation energies beyond the molecular orbital picture, which just appeared as a Perspective in PCCP, we asked whether we can go beyond the MO picture in a systematic way in our understanding of excited states. This endeavour ended up more involved than initially expected but we hope that it presents some interesting physics in a memorable and sufficiently intuitive way.
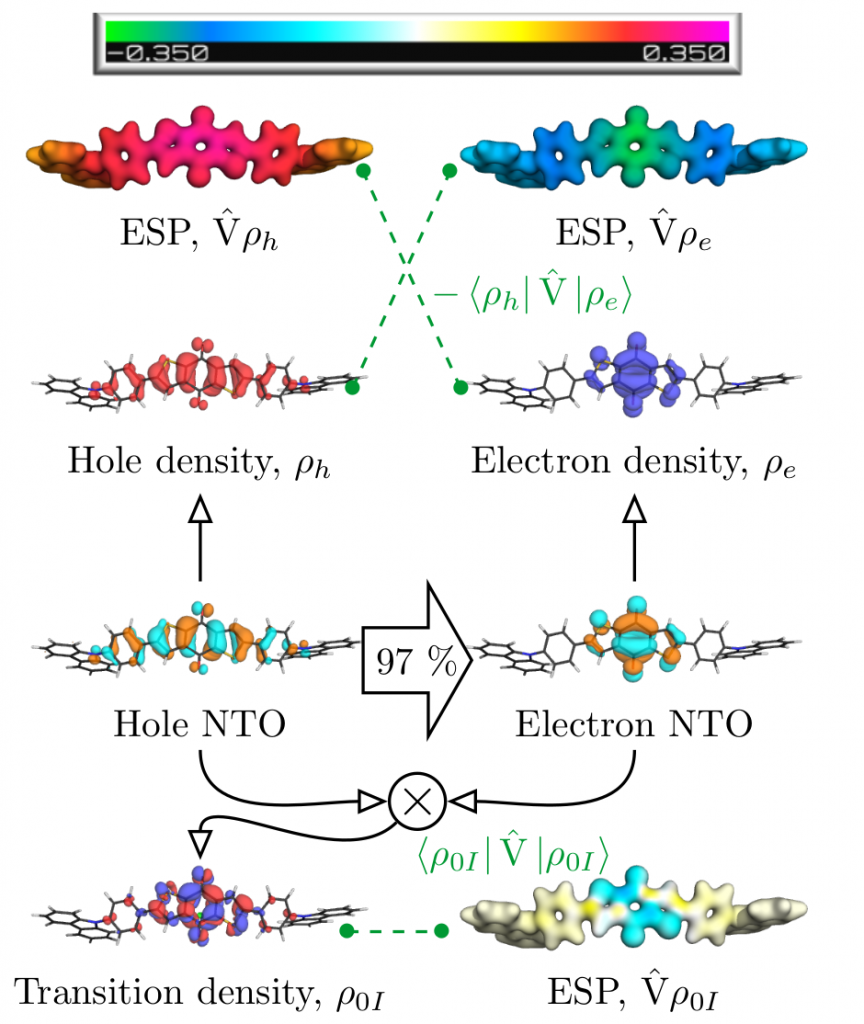
Below, we exemplify the tools developed to study a donor-acceptor-donor system initially developed in JACS, 2014, 136, 18070 and revisited in PCCP, 2019, 21, 10580. The HOMO of this system is on the carbazole donor whereas the LUMO is on the anthraquinone acceptor. The lowest excited state of the system (in gas phase) is not reached via the HOMO-LUMO transition but it is a locally excited state on anthraquinone. Why?
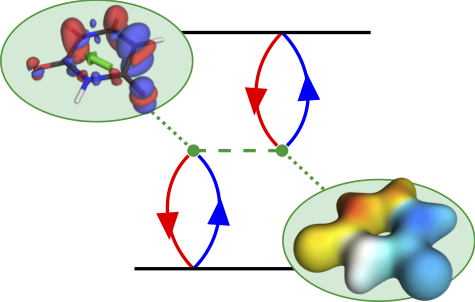
Figure 1: Analysis of post-MO contributions of the first excited singlet state of a donor-acceptor-donor molecule.
The contributions affecting the energy of the first excited singlet state are shown in Figure 1, above. In the third row the natural transition orbitals (NTOs) are shown. These give rise to the density of the excitation hole (red) and the excited electron (blue), shown above. The Coulomb attraction between these two charge distributions now stabilises the excited state. This Coulomb attraction is represented by the electrostatic potentials (ESPs) induced by the charge distributions. The important observation is that these ESPs are non-uniform with stronger contributions in the centre. Thus, the locally excited state is stabilised and an energetic penalty is paid for charge transfer. At the bottom of Figure 1, the transition density is shown, which leads to a repulsive exchange interaction destabilising local singlet states. In the present case the Coulomb attraction dominates and the lowest state is fairly local in nature.
Yesterday, Felix gave a seminar talk at Imperial College, London. Title: Understanding excited states of functional molecules beyond the molecular orbital picture.
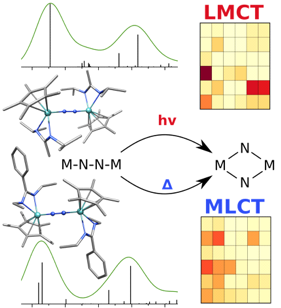
Splitting N2 is a challenging undertaking due to the strong triple bond holding the two nitrogen atoms together. Traditionally, this task is achieved via the Haber-Bosch process but recently interest has shifted to the possibilities of nitrogen splitting via homogeneous catalysis. Computations on the dinuclear transition metal complexes involved can be performed thanks to modern computational hardware and appropriate computer codes. However, the analysis of the excited states involved can become a significant challenge due to the large number of states and difficulties in assigning their character unambiguously. To tackle this problem, we have recently devised a strategy for a detailed analysis of the electronic wavefunctions of the states contributing to the reactive process. The associated paper, led by S. Rupp and V. Krewald from TU Darmstadt, just appeared in the European Journal of Inorganic Chemistry: Multi‐tier electronic structure analysis of Sita’s Mo and W complexes capable of thermal or photochemical N2 splitting.
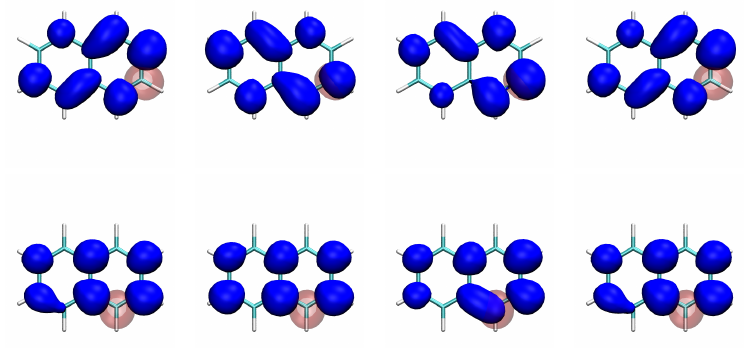
In the tutorial, we explain the process of creating conditional electron densities for visualising electron correlation (ChemPhotoChem2019, 3, 702). The figure below shows a comparison between the ionic and covalent singlet and triplet B3u states of naphthalene.
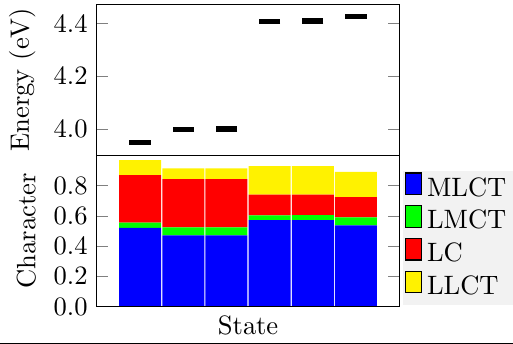
The tutorial also explains the creation of bar graphs for a compact representation of excited-state character (see Coord. Chem. Rev., 2018, 361, 74 and ChemRxiv.11395314). In the picture below, the excited states of an iridium complex are decomposed into metal-to-ligand charge transfer (MLCT), ligand-to-ligand charge transfer (LLCT), and ligand centred (LC) contributions. The lowest six states are all dominated by MLCT character but the presented analysis clearly shows that the first three have enhanced LC character compared to the latter three.
Second, a more extensive paper exploring how far we can use information from excited-state wavefunction analysis tools to understand excitation energies beyond the molecular orbital picture. The energy of a correlated electron-hole pair is derived using diagrammatic techniques and this information is further used for a graphical depiction in terms of different charge distributions and their electrostatic potentials. Doing so turned out not as easy as hoped for but was very exciting. Find more here: Toward an Understanding of Electronic Excitation Energies Beyond the Molecular Orbital Picture by P. Kimber and F. Plasser.
Tomorrow Felix will give a talk at the New Horizons in Materials Modelling 2020 taking place in York. Title: A toolbox for analysing structure-property relationships in functional molecules interacting with light.
Privacy & Cookies: This site uses cookies. By continuing to use this website, you agree to their use.
To find out more, including how to control cookies, see here:
Cookie Policy