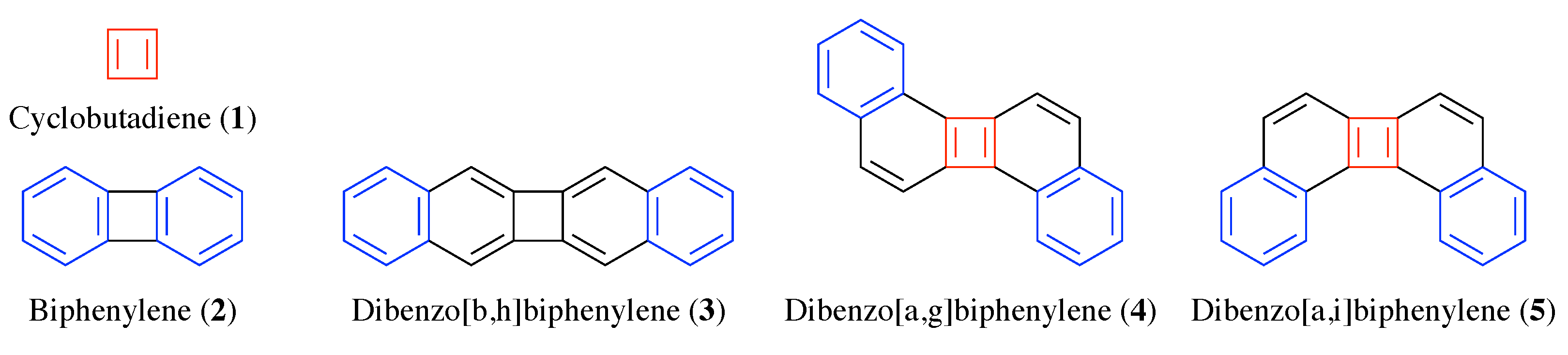
Can you sort these molecules according to increasing triplet excitation energies?
Some basic considerations might suggest that energies go down as the size of the molecule increases. But this is incorrect. The decisive feature of these molecules is their ground-state antiaromaticity along with their potential for excited-state Baird aromaticity. Triplet excitation energies increase sharply going from 1 (0.1 eV) via 2 (1.9 eV) to 3 (2.6 eV). This can be understood in the sense that antiaromaticity is blurred as the molecule becomes larger.
More strikingly, when going from 3 to 4 or 5, the energy drops again dramatically down to 1.0 eV. This effect is explained following Ayub et al. by the simple fact that these molecule possess resonance structures with simultaneous quartets and sextets.
A new study led by J. Lachner from the Helmholtz-Zentrum Dresden describes a method for detecting 26Al via Ion-Laser Interaction Mass Spectrometry using a particle accelerator.
Quantum chemical calculations highlight the different energetics of 26MgO and 26AlO, which are separated with high specificity despite being isobars.
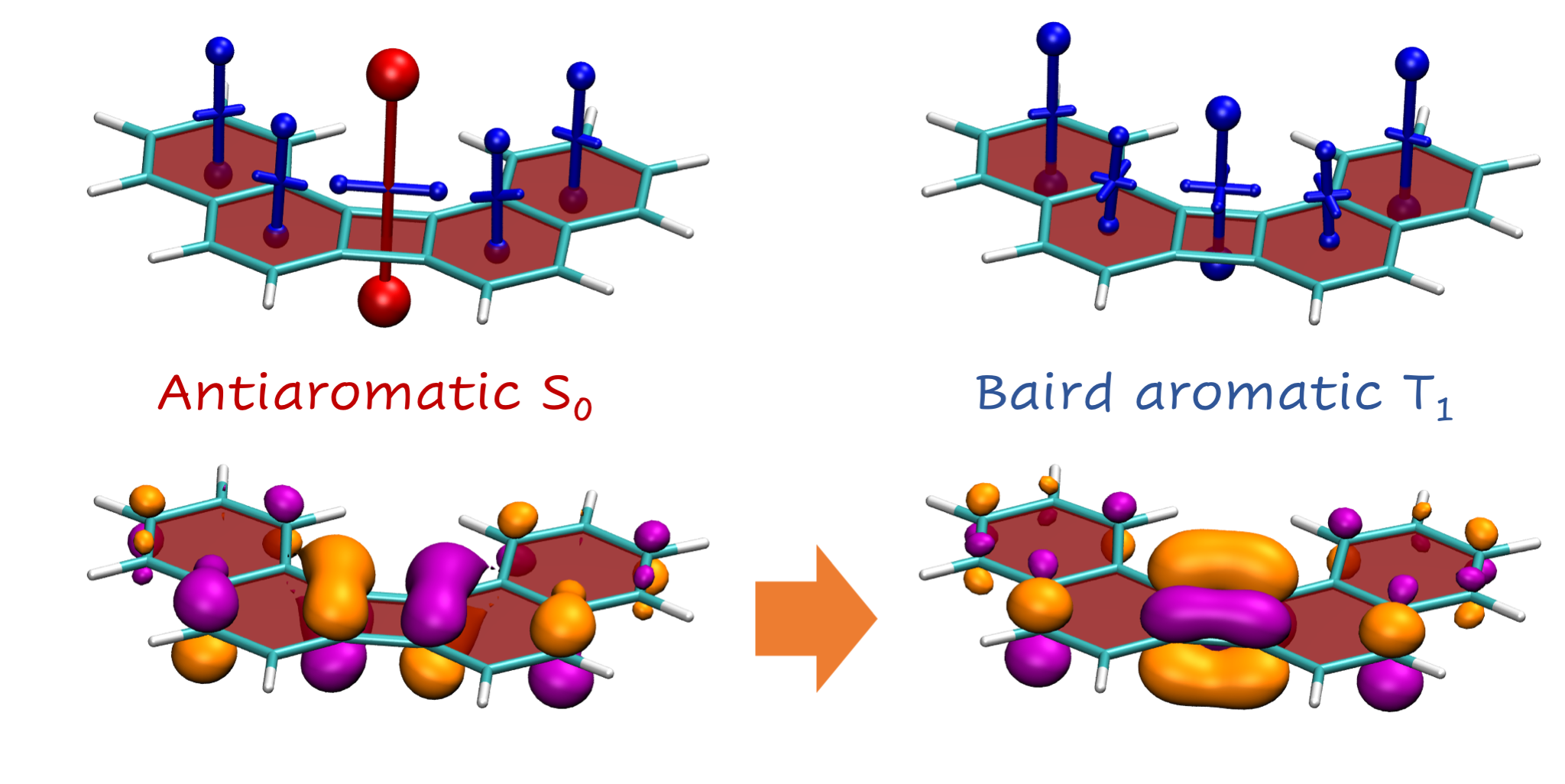
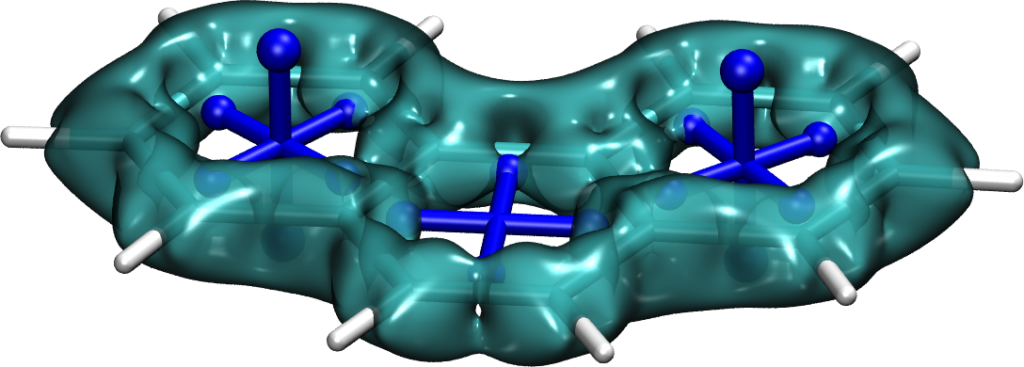
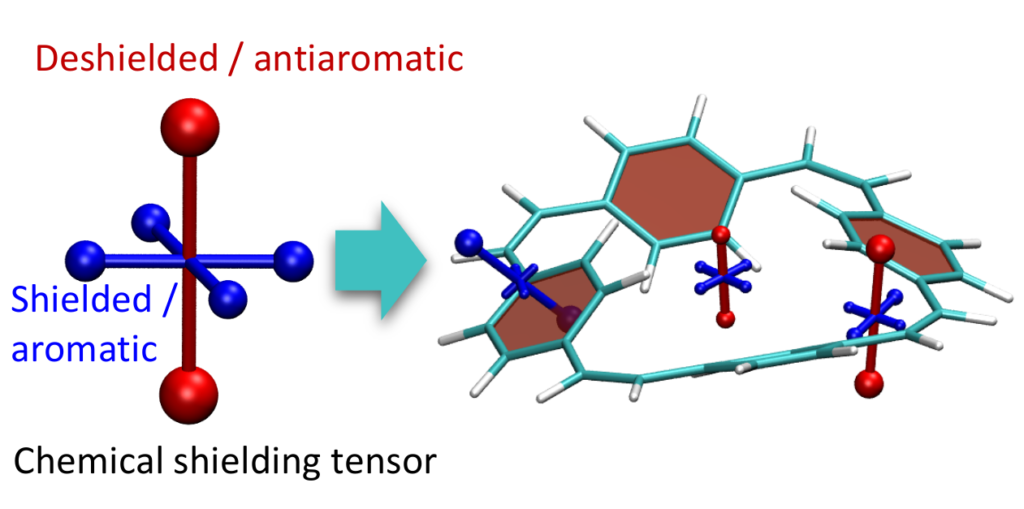
Aromaticity is a ubiquitous yet elusive concept in chemistry and chemists have spent a great deal of effort on developing methods to quantify and visualise aromaticity. One particularly popular method is the nucleus independent shift (NICS), which can be seen as a virtual NMR experiment carried out within a conjugated ring to evaluate the enhanced chemical shielding induced by aromatic ring-currents. Strikingly NICS also allows to quantify antiaromaticity, as this induces a net deshielding effect within the ring. NICS provides a powerful quantitative aromaticity criterion but the main challenge for its graphical representation is that the chemical shielding is a 3×3 tensor, which is difficult to visualise with the existing methods.
Therefore, we have developed a new method for the visualisation of chemical shielding tensors (VIST), which provides a local representation of the shielding tensor along with the molecular structure. The method, thus, allows to probe local aromaticity along with the underlying anisotropy of the shielding. The method is described in the preprint “3D Visualisation of chemical shielding tensors to elucidate aromaticity and antiaromaticity” available on ChemRxiv.
Within the preprent we exemplify the main concepts in the benzene and phenanthrene molecules and continue by studying
the interplay of ground state antiaromaticity and Baird triplet state aromaticity in the potential singlet fission chromophore cyclobuta[l]phenanthrene,
local aromaticity in the neutral formally antiaromatic ground state along with global aromaticity in the doubly reduced state of paaracyclophanetetraene,
Privacy & Cookies: This site uses cookies. By continuing to use this website, you agree to their use.
To find out more, including how to control cookies, see here:
Cookie Policy