Our work has been accepted as cover art for Chemical Science. The figure shows an inositol phosphate binding to a lanthanide receptor molecule inducing a fluorescent respose.
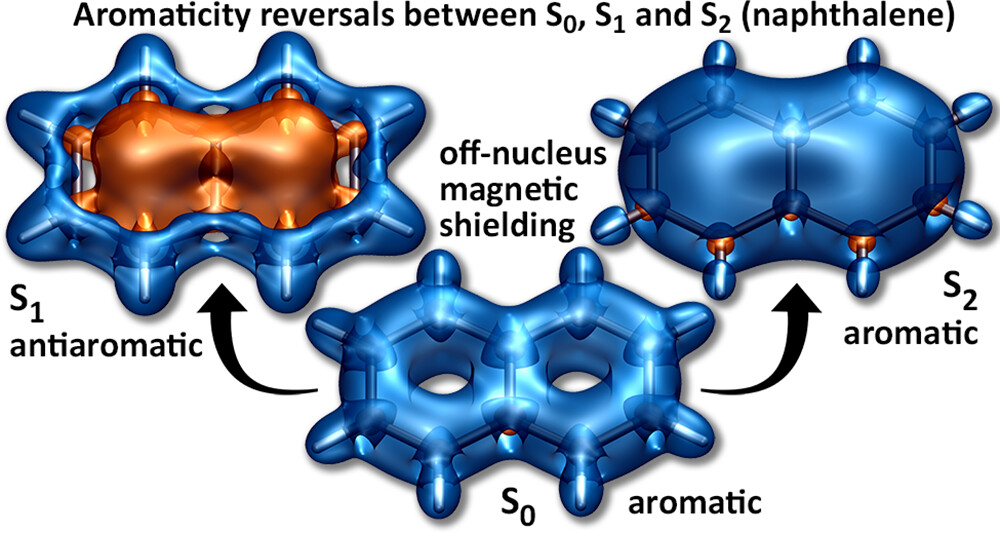
A recent JPCA article by Karadakov and Al-Yassiri highlights the differences in singlet and triplet aromaticity in naphthalene. To me this paper contains several striking observations:
The singlet HOMO/LUMO transition (S2, 1La) is shown to be strongly aromatic whereas the triplet HOMO/LUMO transition (T1, 3La) is antiaromatic. Does this mean states reached by the same kind of orbital transition behave differently depending on their spin-multiplicity?
The aromatic S2 lies above the antiaromatic S1 even though S2 is the HOMO/LUMO transition. Does this mean that singlet antiaromaticity is actually a stabilising effect?
We have discussed the excited states of naphthalene from an entirely different viewpoint in a recent J. Chem. Theory Comput. article. It would be fascinating to combine the two viewpoints.
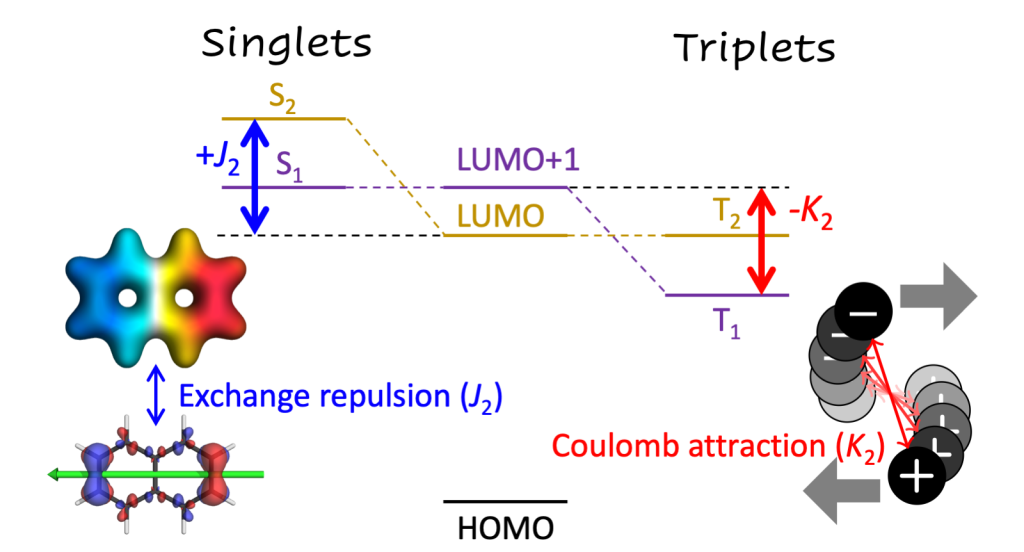
We just posted a preprint discussing a question I have been wondering about for a while: Why is the lowest excited state of a molecule not always the HOMO/LUMO transition? More generally we show how singlet and triplet state energies are affected in different ways by post-MO energy terms.
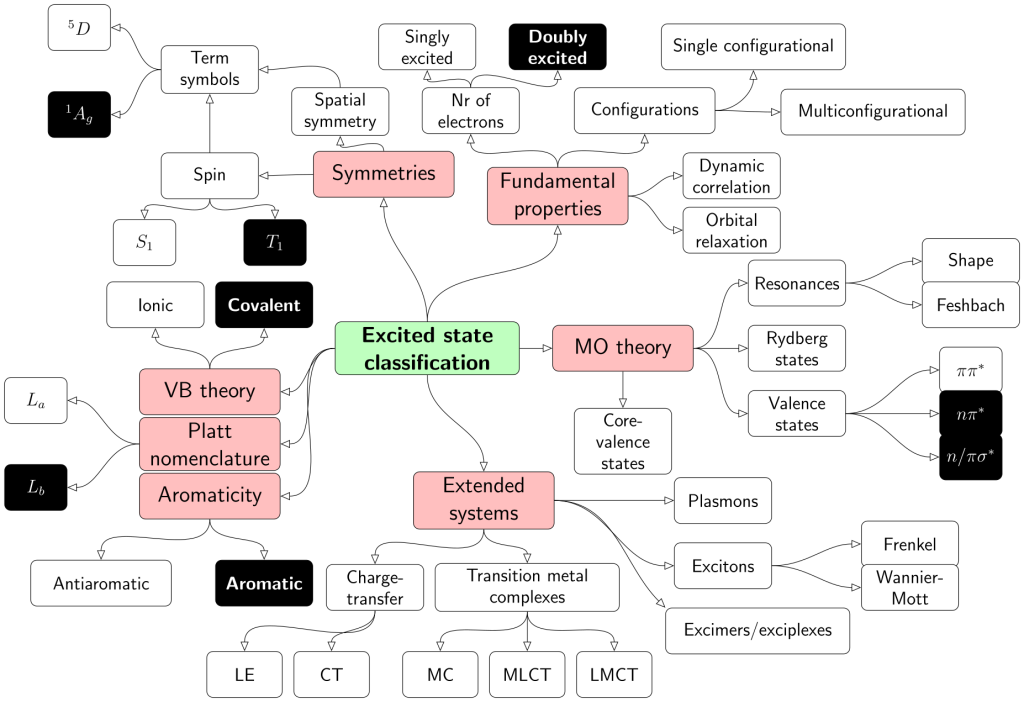
In this chapter we explore the various ways in which excited states are classified, that is, according to
the molecular orbitals involved,
valence bond resonance structures,
spatial and spin symmetry,
more fundamental wavefunction properties (double excitations, correlation, etc),
excited-state aromaticity, and
delocalisation and charge transfer.
The map below shows the different classes and highlights the multitude of ways that are used to discuss excited states in the literature.
It is the purpose of this chapter to discuss all these types of states, covering the mathematical and physical background as well as the consequences to spectroscopy and photochemistry.
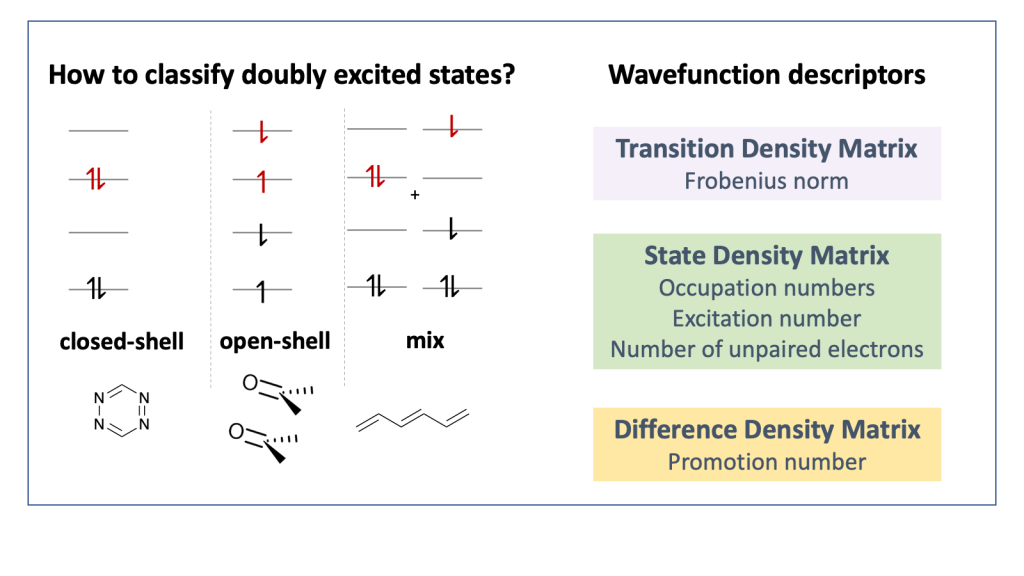
The topic of doubly excited states has been discussed quite controversially in the literature over the last couple of years, see for example JACS, 139, 13770 (2017) and JCTC14, 9 (2018), and it is often disputed whether to classify a state as doubly excited at all. To contribute to this discussion we worked on the development of a physically motivated definition of doubly excited character based on operator expectation values and density matrices, which works independently of the underlying orbital representation. We hope that this approach will provide new understanding on these issues.
The authors discuss the best modern methods for running a DFT computations – a guide through the jungle. For me the main conclusions regarding functionals are:
The composite method r2SCAN-3c offers great cost-benefit a ratio and we are starting to adopt it as a default method for ground-state optimisations. (This should be taken with a grain of salt, since it is the authors’ own method, but the arguments are sound.)
If you want to go beyond r2SCAN-3c, you have to try really really hard, as in range-separated hybrid meta-GGA with a triple/quadruple-zeta basis set. There is not really any reason to ever use double-zeta basis sets – better use a compound method.
This is a flexible position allowing you to apply state-of-the-art quantum chemistry along with sophisticated analysis methods. The goal is to develop new rules for designing functional materials going beyond the frontier orbital picture. The work will apply rules developed in PCCP, 22, 6058-6080 (2020) in connection with experimental partners.
This is a flexible studentship that can be adjusted to your needs and interests. Please apply by February 2023 if this interests you.
Today, Felix will present at the School on High-Performance Multilayer Molecular Dynamics Approaches in Madrid. He will talk about classification and analysis of excited states in the morning, and present the practical use of TheoDORE in the afternoon. Please, see the official web page for online joining instructions.
Kasha’s rule states that fluorescence generally occurs from the lowest excited singlet state (S1). Exceptions to this rule are usually associated with a metastable S2 state that is separated from S1 not allowing for interconversion. In a recent article we outlined a different mechanism for non-Kasha fluorescence: If S1 and S2 are very close in energy, then S2 is populated in a dynamic equilibrium following Boltzmann statistics. This effect is particularly pronounced if there is a large amount of vibrational excess energy following excitation into a high-energy absorption peak. The full story, “Non-Kasha fluorescence of pyrene emerges from a dynamic equilibrium between excited states” was just published in J. Chem. Phys.
Privacy & Cookies: This site uses cookies. By continuing to use this website, you agree to their use.
To find out more, including how to control cookies, see here:
Cookie Policy