A recent study, lead by Florian Glöcklhofer from Imperial College London, explores the effect of methoxy and thiomethyl subtitutions on a formally antiaromatic macrocycle. The corresponding paper “[2.2.2.2]Paracyclophanetetraenes (PCTs): cyclic structural analogues of poly(p‑phenylene vinylene)s (PPVs)” is available via Open Research Europe, 1, 111, 2012.
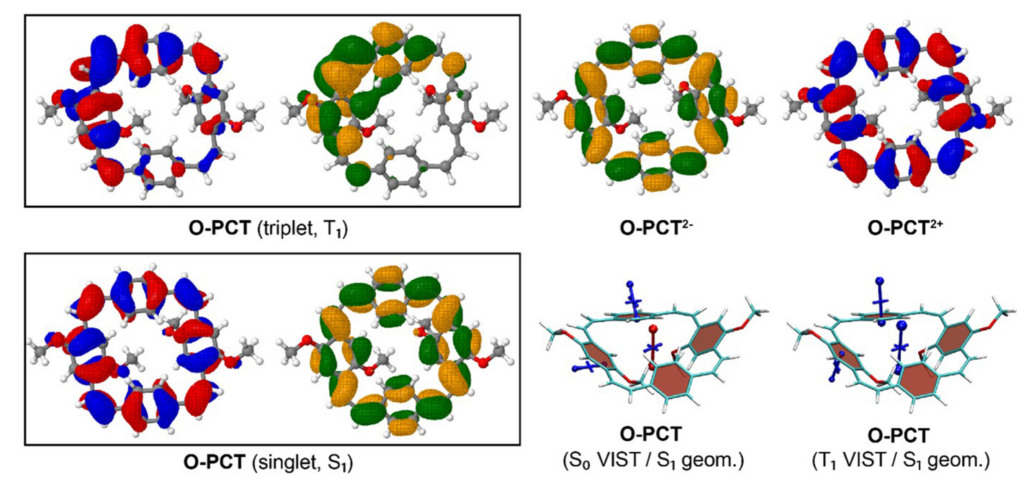
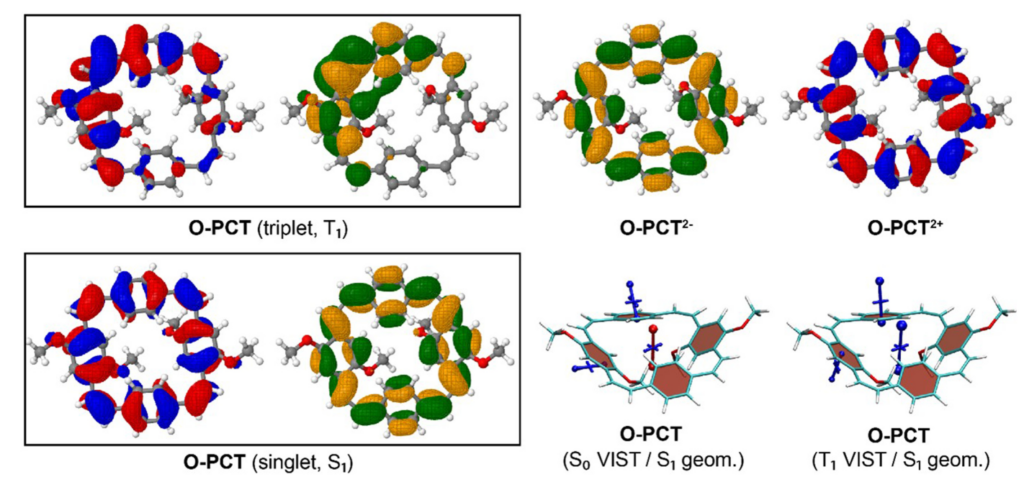
The above figure compares the orbitals and aromaticity descriptors for different charge and spin states. Importantly, the symmetry is broken in the T1 state, inhibiting Baird aromaticity. By comparison, the symmetry is retained for the neutral singlet, dianion, and dication states all of which exhibit aromaticity.