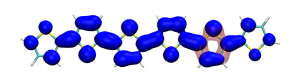
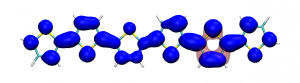
Version 2.0 of the TheoDORE wavefunction analysis package has been released, download below. The two main features of TheoDORE 2.0 are the computation of conditional electron densities and compatibility with python3.
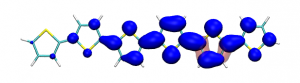
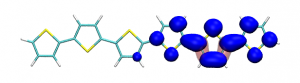
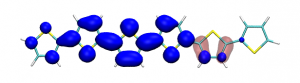
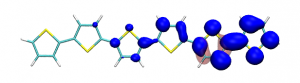
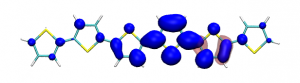
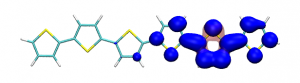
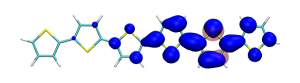
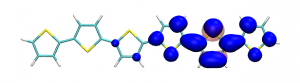
Conditional electron densities can be used for the visualisation of excited-state electron correlation, see ChemPhotoChem (2019). Below, the application of this method to a PPV oligomer is shown. Here, the probe hole (red) is always fixed on the terminal phenyl ring and the different shapes for the conditional electron density (blue) for the first six excited states is observed. One can see that for the different states the electron is either repelled, attracted or unaffected by the hole.
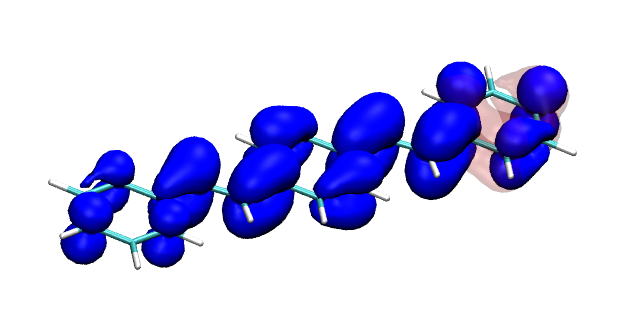 S1
S1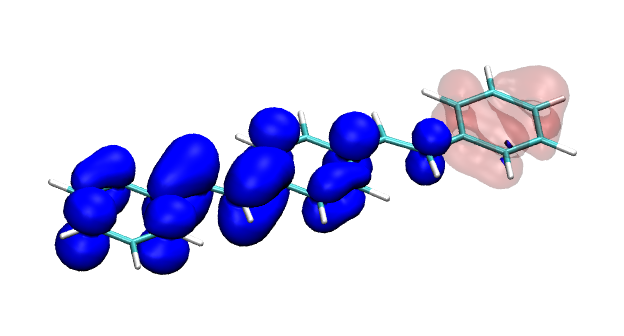 S2
S2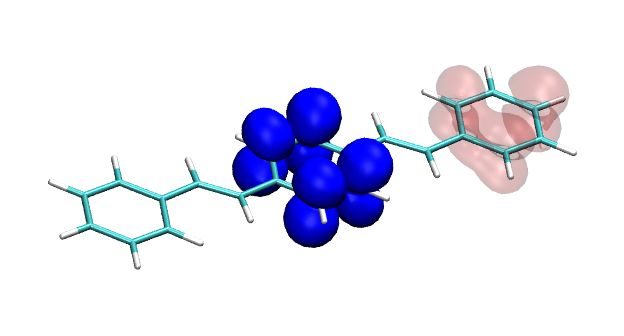 S3
S3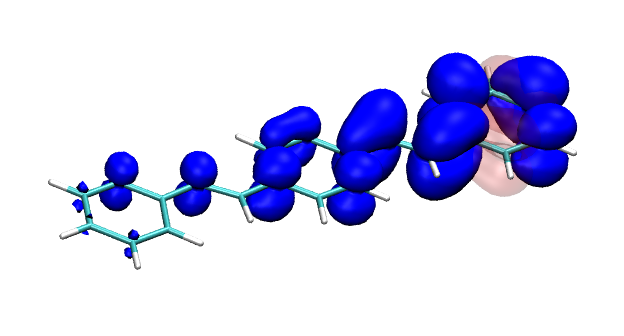 S4
S4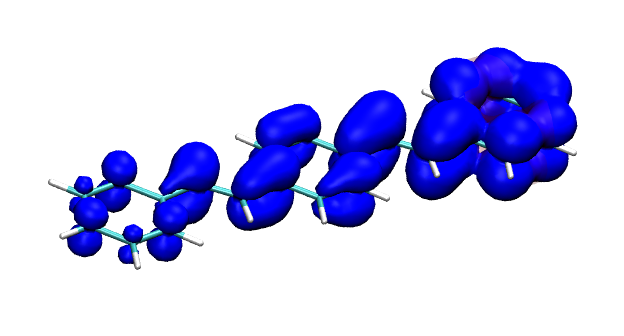 S5
S5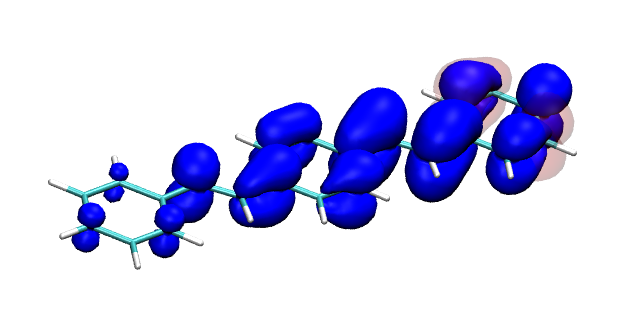 S6
S6